




欢迎观看强生线上论坛!


-
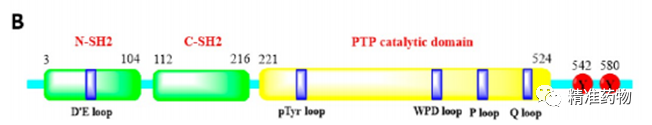
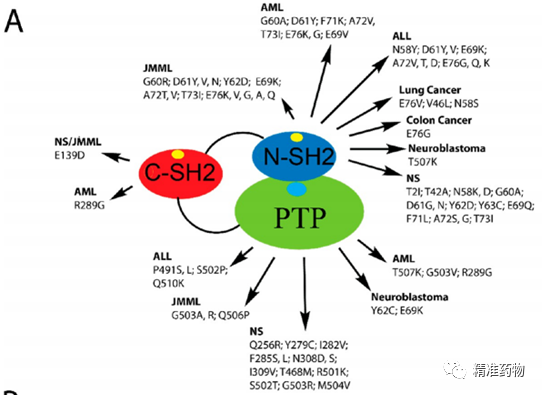
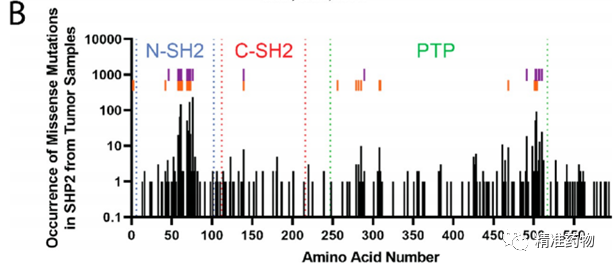
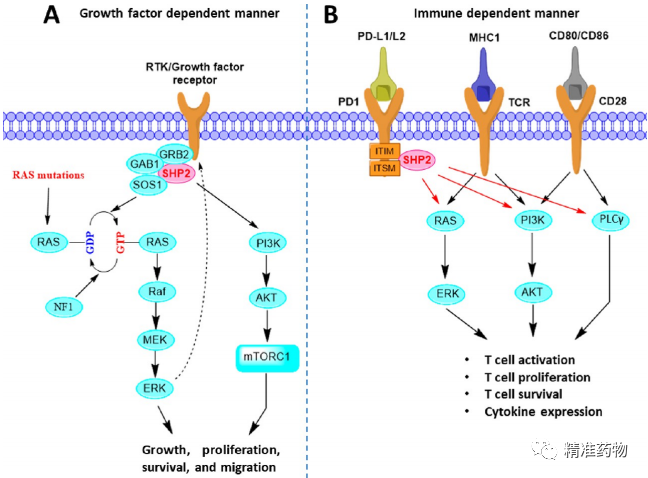
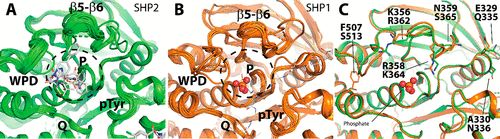
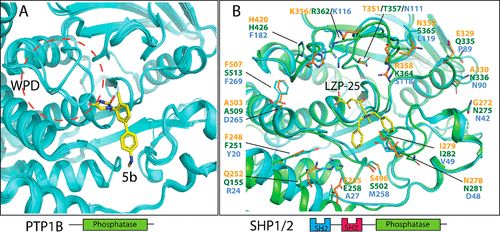
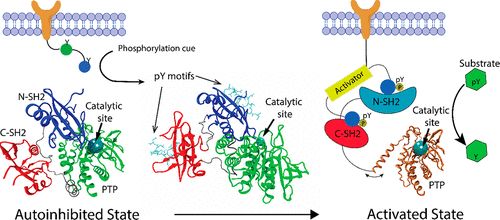
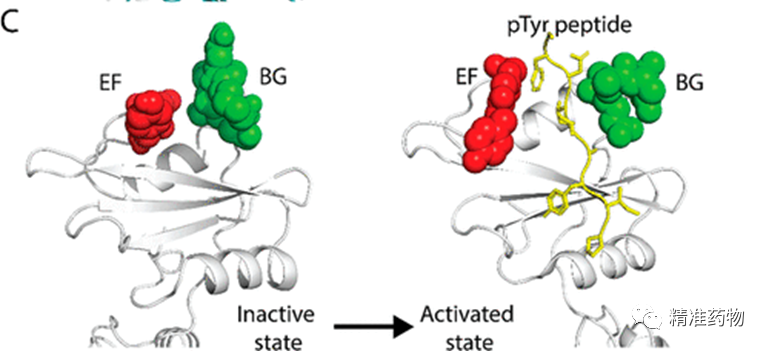
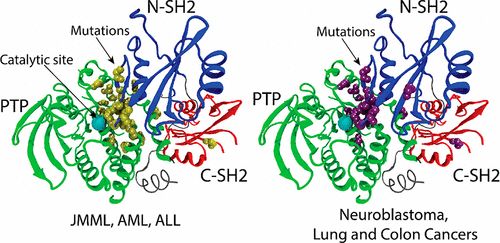
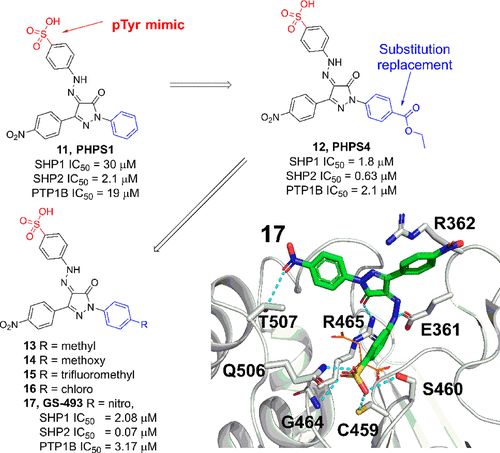
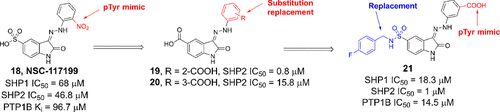
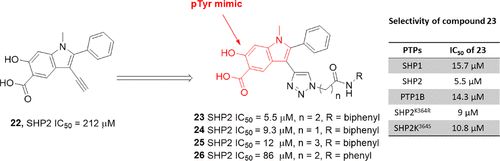
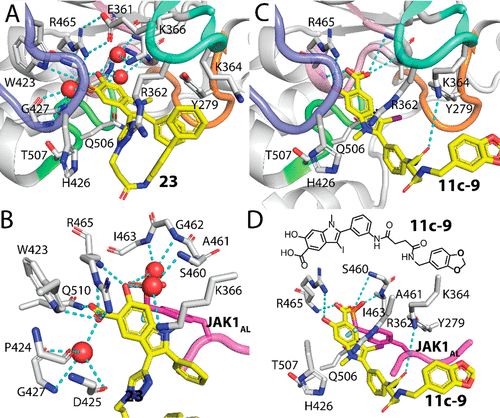
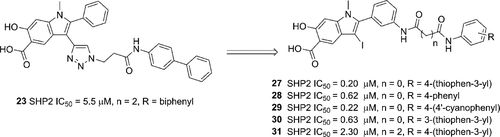
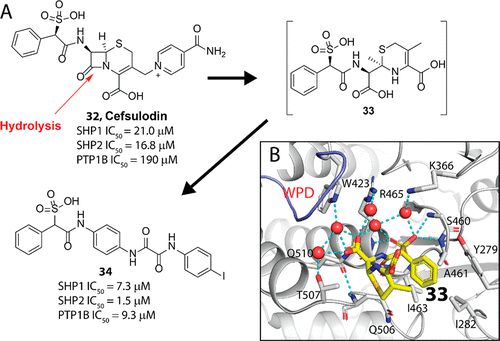
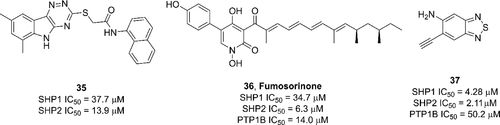
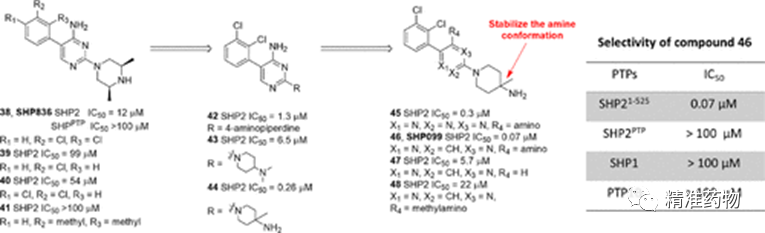
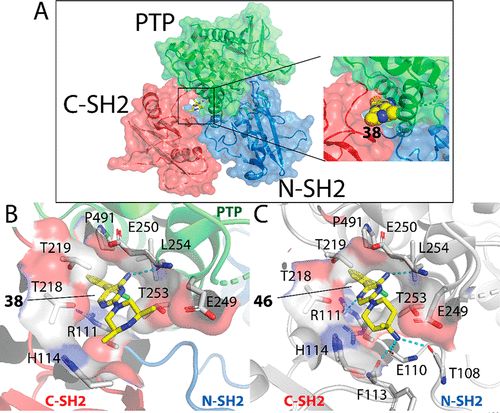
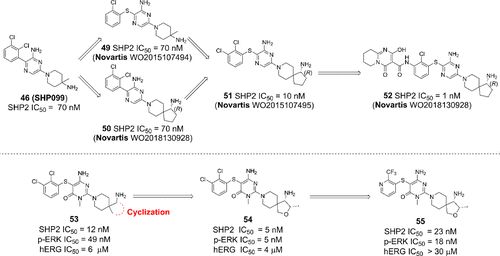
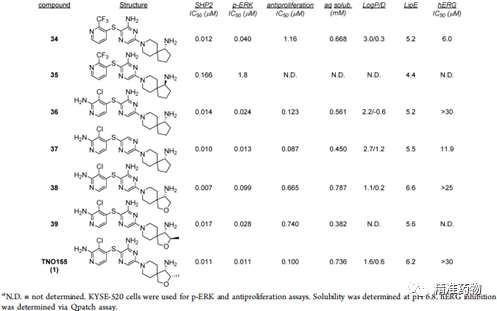
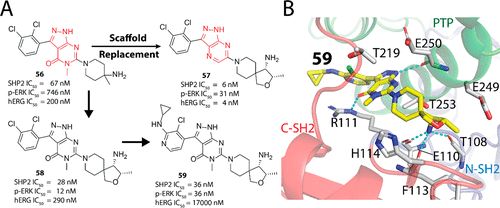
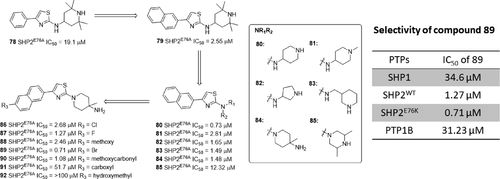
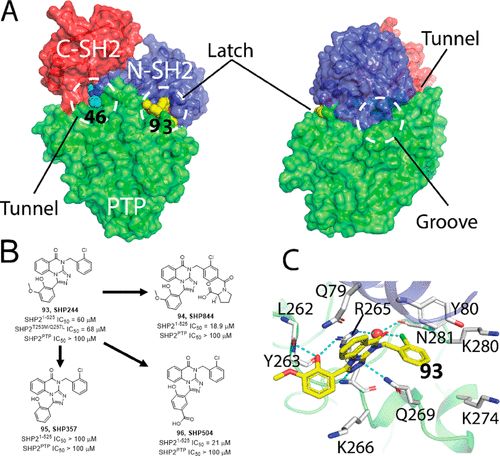
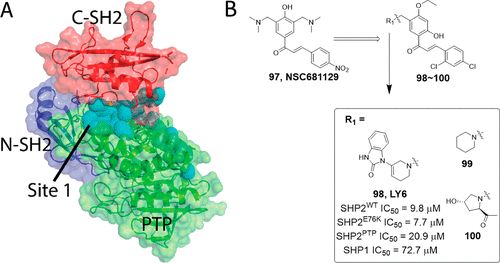
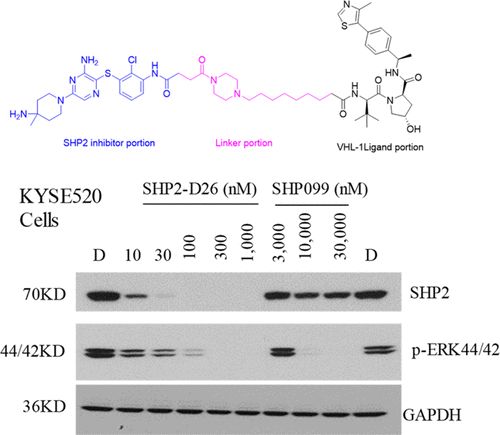
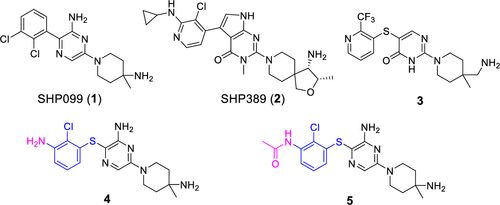
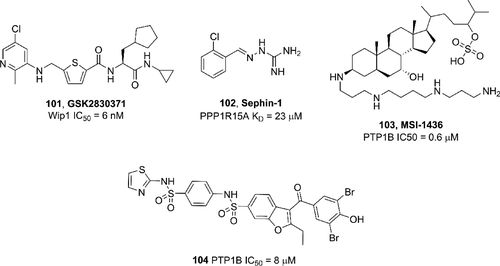
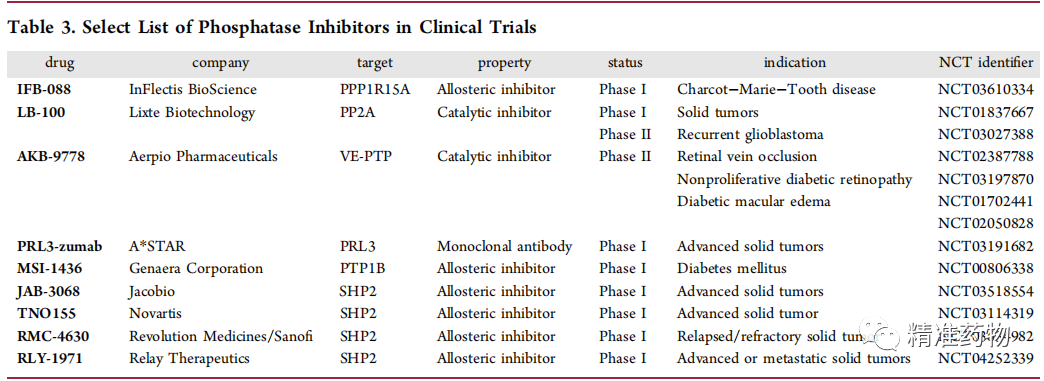
补充:激活SHP2构象含有pTyr的蛋白质主要包含三类:免疫抑制受体,支架蛋白(Grb2相关的结合蛋白[Gabs],成纤维细胞生长因子受体底物[FRS],胰岛素 受体底物[IRS])和受体(受体酪氨酸激酶[RTKs],细胞因子受体)。【European Journal of Medicinal Chemistry,Volume 190,2020,112117,ISSN 0223-5234.】

– END –


 点击直达更多精彩!
点击直达更多精彩!发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 