
上次第一期发完之后,圈内很多朋友关注我们,很高兴得到大家的认可。这次咱们继续,前面铺垫的话就少一点,多一些枯燥的数据解读。
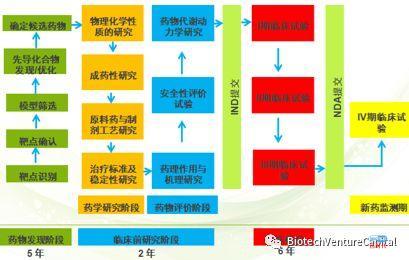
想做好创新药项目的投资,必须要熟悉新药的开发流程。上图是新药研发的大概流程图,从上图可以看出,新药的开发可以简单分为药物发现阶段、临床前研究阶段和临床研究阶段三大阶段。本文将重点对后面两大阶段分别进行介绍:
临床前研究阶段(IND前)
对照药物研发流程图,处于该阶段的项目已经成功跨越了药物发现阶段,药物作用靶点明确且经过苗头化合物、先导化合物筛选阶段,属于众多化合物中活性最适合、DMPK、体内体外实验验证后最中意的一个或几个,定为临床前候选化合物(PCC)。
随后,针对该化合物会开展大量的实验研究,包括CMC(化学制造和控制),体外(酶学/细胞学)或者体内(动物)药效学(PD),药代动力学(PK),毒理学,制剂开发等多个环节。大多数创新药企会参照VIC模式(风险投资+知识产权+CRO公司)进行新药研发,将很多实验外包给行业内知名的CRO公司,对于个别项目的自己做实验的数据要格外注意。
化学、制造和控制
(CMC)
临床前候选药物的确定标志着新药项目正式进入临床前研究阶段,那么API如何制备则是新药研发第一步需要考虑的问题,因为它是药物研究和开发的基础,是药物研发的起始阶段。它决定了能否为后续药物研发过程中药理毒理、制剂、临床等研究提供合格的API,也影响是否可以通过对工艺全过程的控制来保证生产工艺的稳定、可行,同时也制约着是否可以为上市药品的生产提供符合要求的原料药?
当然,这是一个不断改进、完善的过程,始终贯穿于新药研发的不同阶段,从最初的药物发现到药物上市,新药研发企业不可能在最开始就把它做到最好,这也是很不现实的。
通常情况下,第一批提供的API主要用于毒理学研究,要求是越快越好,成本并不是主要考虑因素。因此,只要合成路线能够满足毒理学研究用量,工艺研发部门就会采用。
但随着项目的推进,工艺部门会继续优化设计合成路线,以满足从临床I—III期试验用药与商业化的需求;同理,制剂部门首先也会以最简单的形式给药,完成毒理学研究,然后不断完成处方工艺研究,开发出适合商业化的制剂工艺。

关注后续的生产工艺和开始时相比是否优化改进,目标化合物需要经过几步合成路线完成?是否包含特殊或者严格苛刻的合成条件(无水无氧,高温,高压,剧毒等)?每一步的提纯处理是否容易操作,最终实验得率如何?工艺是否稳定,最大生产规模可以达到的级别临床试验中采用的剂型是什么?药物稳定性如何等等。以上这些问题都是我们所关心的。
在尽调过程中,CMC并不算是工作重点,相对来说,了解该化合物结构思路设计来源更为重要。是在已上市药物基础上基于经典药化设计改造还是其他(HTS, FBDD, DEL, virtual screening)?若为改造,和原来化合物的优劣势比较在哪里?
药代动力学
(Pharmacokinetics,PK)
通俗的说,药代动力学研究的是身体对药物做了什么;正式的说,它是通过体外和动物体内的研究方法,揭示药物在体内的动态变化规律,获得药物的基本药代动力学参数,阐明药物的吸收、分布、代谢和排泄(ADME)的过程和特征。
创新药一般选用两种或两种以上的实验动物,其中一种为啮齿类动物,另一种为非啮齿类动物,最好与药效或毒性研究所用动物一致,并且是与人代谢性质相近的动物。
这部分是我们在项目尽调过程中的重点内容,我们会关注拟推化合物PK试验设计和所获得的药代动力学参数。
比如:实验动物的种属,是否雌雄各半,样本量是否符合统计学要求,所得数据是否有性别差异?实验动物给药剂量分为几组,具体剂量选择的依据是什么,药物在动物体内的动力学过程是否属于线性?是否采用体外吸收模型(如Caco-2模型)研究药物吸收机理及影响因素,食物是否影响药物的吸收,药物的生物利用度是多少?是否比较药物在动物与人之间的血浆蛋白结合率差异?
若为治愈中枢神经系统的药物,需要考虑它能否很好的穿透血脑屏障(BBB)?
事实上,所有的大分子和98%以上的小分子药物都不能穿越完整的BBB,而且BBB的药物外排泵(如P-糖蛋白),能将某些药物主动排出脑组织。那么,如何判断一个化合物能够很好的穿透BBB进入脑组织而发挥治疗作用呢,这就成为我们尽调核查的关键点。
一般项目方会提供一些比较单一的实验数据,我们认为应该从多个维度的实验多个方向去验证:
1)大鼠单次口服给药后的血浆和脑脊液中不同时间点的药物浓度;
2)大鼠单次口服给药后的血浆和脑组织中不同时间点的药物浓度;
3)大鼠单次口服给予同位素标记的药物后的血浆和脑组织中不同时间点的药物浓度。最后三个独立的实验都得到关键数据——稳态时脑血分配系数(K p,uu),它可以来评估药物进入CNS(中枢神经系统)的能力。
观察药物在动物和人肝微粒体内的代谢情况。如药物的代谢是否具有明显的种属差异,明确药物对细胞色素P450酶的诱导或抑制作用,了解药物可能在代谢方面存在的相互作用。
对于临床需长期给药的药物,更要注意重复给药是否有蓄积倾向?
最后,药物半衰期T1/2,达峰时间Tmax,最大血药浓度Cmax,血药浓度-时间曲线下面积(AUC),表观分布容积V等多个参数也需要我们仔细去考量。
(Pharmacodynamics,PD)
正式的说,药效学是研究药物对机体的作用及作用机制。通俗的说,药效学关注的是药物对身体做了什么。
动物药效学实验是临床研究的基础,药物在无动物实验数据的前提下,应用到临床,势必会对人体造成无法想象的伤害。所以,新药研发会首先通过体内或体外药效学实验来提前预判药物对某种疾病是否有效,安全窗范围是多少,有哪些不良反应发生?
药效学实验采用随机,对照,重复的原则开展,在药物早期研发阶段,我们会开展大量分子和细胞水平的研究,比如与受体的结合、对激酶的活性(包括特异性、可逆/不可逆性、时间依赖性等)以及对不同肿瘤细胞系细胞生长抑制的活性与靶向性等。
随后,即开展动物药效学研究,预测临床适应症、量效关系(有效暴露量、临床剂量范围等)、时效关系(临床给药周期)、给药途径、药效指标等,分析影响药效因素,帮助后期设计临床试验,都具有重要意义。
所以,这也是我们新药项目尽职调查的重点内容。
以肿瘤药物为例,我们需要关注药物对目标适应症做过哪些药效学试验,是否涵盖体外和体内试验?体外药效学实验中采用多少种细胞系,是否是业内公认或者大量文献证实的细胞株?是否设置阳性对照和空白对照组,采用何种方法检测和计算半数抑制浓度(IC50)?是否在选择肿瘤细胞系时考虑到细胞的生长和增殖速率?试验是否重复至少一次?
这里要插一个题外话,也是我们自己的思考,是否应该根据靶点生物学作用原理的不同,而选择不同的作用评价指标?比如,BTK靶点是要强烈抑制封闭这个通路,所以要达到IC90这个效果比较好,IC50就不行。其他导致细胞凋亡的作用通路上的靶点可能IC50就OK了,用不着IC90。所以按照这个推导是不是CDK4/6靶点的就需要IC90这个指标更适合?因为这个靶点的作用是抑制细胞分裂在G1期,并不是杀伤细胞或者导致细胞凋亡。

而体内试验是用于进一步考察药物对特定类型肿瘤细胞的杀伤或抑制作用,探索药物产生药效作用的给药剂量、途径、频率和周期等。此时需要关注体内试验采用何种动物模型(CDX/PDX),肿瘤移植部位(皮下/原位),检测指标和评价指标,统计学结论,以此来判断化合物的有效性。
虽然,该药效学实验遵从既定原则设计完成,但其最终得到的实验结果也具有一定的局限性。因为人和实验动物之间存在种属差异,临床疾病和动物模型间也存在着差异,动物反应和人体表现之间也是有一定的差异。所以,在新药开发历程中,流行着这样一句话:在小鼠体内成功治愈肿瘤已经有千万次了……
毒理学研究(Toxicology)
通常情况下,新药研发的临床前和临床阶段都需进行药物毒理学研究,而临床前毒理研究包括急性毒性、重复给药毒性、安全性药理、特殊毒性(遗传毒性、生殖毒性、致癌性)、毒代动力学、其他毒性(刺激性、过敏性、溶血性),以及免疫毒性和依赖性等。
其研究目的是发现药物的毒性反应的症状、出现持续和结束的时间、无毒性反应的剂量水平、毒性反应的剂量与药效剂量的间距(即治疗窗或安全范围)有多大、毒性反应的性质与可逆性等。
为了加速新药能及早验证是否有疗效,尤其是对一些抗癌药,有些耗时费钱的毒理实验(如致癌性、生殖毒性)是可容许在临床试验阶段再进行。毒理学研究也是选择两种实验动物(啮齿类+非啮齿类)进行研究,同药效学与药代动力学相一致。该部分也是属于项目尽调的重点内容,其中一般药理学是研究药物对于心血管、呼吸、中枢神经系统的影响。
比如在心血管方面,我们会非常关注HERG IC50值,以此来判断它的心脏安全性。那么HERG IC50是否有一个明确的界限来确定药物的心脏毒性,比如大于30µM就一定没有心脏毒性吗,或者小于10µM就一定有心脏毒性吗?这个不是绝对的,有大于30µM的药后来也有产生毒性的,有小于10µM却能获批上市成药的。所以这个HERG IC50是相对的参考值。大家可能就会迷惑了,既然都有可能,那么临床前怎么来判断和分析HERG值的指导意义呢?这个展开来讲还是很多东西,以后我们会针对HERG写个专题,大家保持关注即可。

其次,我们也会通过以上毒性实验来判断多大剂量/暴露量在动物上产生毒性作用?多大剂量/暴露量在动物上未产生毒性作用?所用的动物相关模型是否能预示人体毒性?毒性反应的表现和持续时间?毒性反应是否可逆?毒性靶器官或者靶系统是什么?所表现出的毒性是否是该类化合物可预期的?是否产生毒性代谢物?最后,我们也是通过以上毒性实验得到MTD值,NOAEL值关键性数据,为后续进入临床阶段I期临床试验的初始给药剂量提供依据。
制剂开发
制剂开发是药物研发的一个重要环节。早期制剂研究并不需要完整的处方开发,所有研究围绕毒理学研究和一期临床时方便给药即可,目的是将候选药物尽快推向临床。该部分内容所以也不会是项目尽调核查的重点,但随着项目推进,给药方式和处方研究就越来越全面。
比如,有的药胃肠吸收很差,就需要开发为注射剂。有的药对在胃酸里面会失去活性,就需要开发为肠溶制剂。有的化合物溶解性不好,也可以通过制剂部分来解决这个问题。
自己实验室做的数据
有一些特别早期的项目,因为创始人是高校的教授,很多临床前实验并没有委托CRO公司开展,而是在自己高校的实验室依靠数届学生共同完成,这样既可以节省大量的费用,有廉价研究人员可以使用,也可以按计划推进实验进度,可谓一举两得。
当然,没有任何人会认为这种方式是不可取或者不正确的。但是对于这类新药项目,作为投资人,在项目尽调核查时,除了上面列举的重点之外,我们一定要重视原始数据朔源,查看原始实验记录,以药效学实验为例,可从以下维度进行:实验动物的选择(数量/体重/性别),动物模型的建立(时间/方法/评估),成功建模动物的分组(原始评价指标监测与记录),动物实验流程(空白对照组/阳性对照组/实验组,给药方法,给药周期,溶媒选择等),实验结果记录与解读,统计学结果,最终实验结论等等。
只有这样,才能全面真实地了解项目最初的状态。之前我们尽调过一个项目,但是在尽调中却发现:1)也没有完全遵从既定的实验方案,存在多处严重方案违背且没有合理解释;2)原始实验记录保存不完整。所以,实验数据展示的效果就值得怀疑了,因为实验条件发生了变化。
前面这些内容都统称为临床前研究,是药物开发的第一阶段。临床前各个实验的步骤可不是严格按照上述这个顺序展开,而是一个相互包容、相互协调的关系。比如,原料药工艺研发部门,完成毒理批样品合成后,就必须立即开展合成路线的选择,开发新的合成工艺,提供足够量的原料药以满足制剂部门制剂研究用原料药和后期开展I期临床用药的需求。因此,项目推进是否顺利,就看各专业间的协调与配合是否密切了。
临床研究阶段(IND后)
当一个化合物通过了临床前实验后,需要向药监部门FDA提交新药临床研究申请(IND),其主要目的是向FDA提供数据证明该药物具备用于早期临床试验的合理性与安全性。
IND申请需要以下三个方面的基本信息:
(1)临床前动物药理/毒理学研究以及任何以往的人体应用经验,可包括在美国以外的资料;
(2)生产信息,包括药物产品的组成、生产工艺、生产商、稳定性和生产控制等信息;
(3)临床试验方案、研究员手册、受试者的知情同意书、伦理委员会(IRB)的批准以及附上任何有关IND法规条例。在美国,FDA对IND审评时限规定为30天,在此期间没有被驳回,那么该新药就可以进行人体试验。当然,对于IND的申请,FDA主要考虑的是安全性,这个大家要心知肚明。

市场上见多了项目,免不了遇到一些项目方吹嘘自己的项目获得了多少美国FDA的IND批准,本来圈内专业的人都是不搭理这茬的。但是有些投资人也愿意听,还拿着这个东西上会说可以证明企业的技术实力,我们只能表示呵呵。
大多数新药项目要完成1-3期所有临床试验,极少数新药项目适用于FDA加速审批的绿色通道,针对某些临床急需的疾病,无药可治的可以在二期临床时用替代终点或中期终点来作为临床终点,进而实现提前上市,上市后继续做临床后续验证。
这个在美国FDA叫Accelerated Approval,中国NMPA(我还是习惯叫CFDA,NMPA太拗口了)以前没有,去年开始学习美国,临床急需的可以提前上市,叫有条件性批准上市。
I期临床试验
简单点说,Ⅰ期临床试验主要是围绕患者耐受性和药代动力学展开,当然也可以按照需要同步考察药效学、食物影响、药物间相互作用等。
这一阶段的临床试验一般需要征集20-100名正常和健康的志愿者(对肿瘤药物而言通常为肿瘤病人,但人数更少),在严格控制的条件下,给不同剂量(随着对新药的安全性了解的增加,给药的剂量也逐渐提高,并可以多剂量给药)的药物试验于志愿者,仔细监测药物的血液浓度、排泄性质和任何有益反应或不良作用,以评价药物在人体内的性质。
同时也要通过这一阶段的临床试验获得其吸收、分布、代谢和排泄以及药效持续时间的数据和资料;以及药物最高和最低剂量的信息,以便确定将来在病人身上使用的合适剂量。
结合大家关注较多的肿瘤领域,I期临床试验尽调核查的数据包括:
1)最大耐受剂量(MTD)或剂量限制性的毒性(DLT)是什么?
2)药代动力学参数及其与药效/毒性间的关系(PK/PD)如何?
3)是否可以为Ⅱ期临床试验的拟定受试人群、推荐剂量和给药方法提供很好的依据?
比如,如果1期临床中的多剂量递增试验,最大给药剂量是100mg,bid,在该剂量下第14天药代动力学结果显示,其中前19小时的血药浓度值都大大超过了1 µM,而该化合物在前期人的全血体外实验中得到IC50 是在1 µM(注意这里是总的血药浓度值,不是游离值),根据药代动力学曲线保守估计,每天24小时中至少有19小时最低药物浓度超过IC50,达到预期的人体有效治疗浓度。这个时候至少可以知道了人体按照100mg,bid连续给药,每天可实现19小时以上达到预期的目标药效浓度。
然后根据该给药剂量下出现的不良事件(AE)等级、数量及系统器官,来判断是否还需要继续递增剂量。如果这个剂量对应的AE都是二级的,连三级都没有。那么对于肿瘤适应症来说给药剂量肯定要继续往上爬,因为还有足够的安全窗去尝试,每递增一个剂量都意味着在二期的时候可以离达到临床终点更进一步。
好了,我们举举反例。
之前有个项目也是在1期开展中,项目方也是宣传他们的安全性有多高,起始剂量就从400mg开始,然后在入组的最开始的三个病人身上服药后, 没有任何明显副作用,病人感觉良好。后来又爬到了900mg,还是很不错。

表面上好像真的很好哦,然后看PK数据,暴露量也不低啊,好像安全窗还蛮高。其实仔细想一下,他这个化合物血浆蛋白结合率是99.7%,游离量非常低,PK数据得出来的血液中的总的血药浓度虽然高,但是游离量太少,经过计算,离目标有效浓度还是有一定距离。所以这个所谓的高剂量如此安全,是假安全,离真正的坡顶还早呢。
二期临床试验的项目我们很少考察,基本上也不在我们的投资范围内,真正好项目进展到了二期临床,估值已经比较贵了,不适合我们的定位,可能适合其他类型的投资机构或者大药企。所以这里就不分享二期临床的尽调重点了。
我们可以提醒大家的是:不要被二期临床试验中部分患者的CR或者ORR的所谓惊艳数据所陶醉,有时候留心下细节,会发现疗效和剂量非依赖性也是一个角度哦,从那个角度看过去也许是另外一番数据的解读。这里就是点到为止,大家自己多琢磨。
好了,第二期我们就讲到这。行文仓促,若有不对或不准确之处,还请各位专业人士给予指正。下期请关注专利尽调。
如果对我们感兴趣,可以加微信:youxuanzibenlifang 或 cfwf1010
以上文字由Biotech venture capital原创,仅为促进讨论与交流,不构成法律意见或咨询建议。 版权所有,违者必究。如需转载,请注明作者和Biotech venture capital原创。
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 
