


专家介绍:何旺骁

博士,西安交通大学特聘研究员,博士生导师。陕西省高层人人才引进计划入选者,西安交通大学“青年拔尖人才”支持计划入选者。主要从事靶向蛋白蛋白相互作用的多肽药物开发与递送。近 5 年内发表 SCI/EI 期刊共 35 篇,其中领域内权威期刊(IF > 8)20 篇、JCR 1 区刊物 21 篇;2018 年至今以第一或通讯作者在 Adv Funct Mater,ACS Nano,Nano Letters,Biomaterials,Chem Eng J,Chem Mater,J Controlled Release,Theranostics 等刊物上发表论文 19 篇(累积 IF > 180),其中 JCR 1 区 14 篇、IF > 10的 11 篇、IF > 15 的 1 篇、ESI 高被引论文 1 篇。申请美国发明专利 2 项,中国国家发明专利 4 项
专家介绍:闫 瑾

工学博士,西安交通大学第二附属医院研究员。曾入选国家博士后创新人才支持计划。从事功能多肽分子开发、多维度精准调控致病蛋白-蛋白相互作用网络的相关研究。主持国家自然科学基金、陕西省级自然科学基金、中央高校科研基本业务项目等,申请发明专利 4 项。近 5 年累积发表 SCI 学术论文 20 余篇,以第一或通讯作者发表高水平研究论文(IF > 8 或细分领域 Q1 区顶级期刊)17 篇,包括:ACS Nano,Adv Funct Mater,Nano Letters,Chem Eng J,Biomaterials,Theranostics,Small,J ControlledRelease,Chem Mater 以及 1 篇入选 ESI 高被引(top 1%),累计影响因子大于 190,H 指数为 12。
点击下方收听作者原声解读
正文
靶向蛋白-蛋白相互作用的抗肿瘤多肽药物的开发与递送
杨文光 1,高汝青 2,闫瑾 3*,何旺骁 1**
(1. 西安交通大学第一附属医院人才高地,陕西 西安 710061;2. 南昌大学医学院,江西 南昌 330031;3. 西安交通大学第二附属医院国家地方联合生物诊断与治疗中心,陕西 西安 710004)
[摘要]蛋白-蛋白相互作用(PPI)网络异常是肿瘤发生的重要原因。近年来,多肽药物由于其独特的优势被广泛用于靶向肿瘤相关PPI 调节剂的开发,并获得了成功。通过对近几年的相关研究进行综述,从靶向 PPI 多肽的设计与应用角度总结抗肿瘤药物的最新研究进展,以期为基于靶向 PPI 的抗肿瘤多肽药物研发提供参考。
蛋白质相互作用(protein-protein interaction,PPI)网络呈现基因产物彼此之间的物理相互作用,是理解细胞生命活动的基础,例如新陈代谢、细胞周期控制和信号转导等 [1]。目前已经通过遗传学、生化或生物物理方法(例如荧光共振能量转移或 X射线晶体学)鉴定许多蛋白质之间的相互作用。尽管 PPI 网络尚不完整且容易出错 [2-3],但是对该系统的研究已被证明对于破译网络中蛋白质结构和功能之间的关系、发现新的蛋白质功能、识别功能一致的模块和揭示保守的分子相互作用模式至关重要 [4-6]。此外,PPI 网络为联系不同的疾病与蛋白质 [7],研究药物-靶点的药理关系提供了强有力的工具 [8-9]。
PPI 是细胞进行生命活动的基础。反过来,这种相互作用的异常改变会扰乱细胞正常的生命活动,并导致包括癌症在内的许多疾病 [10]。PPI 界面的突变通常与蛋白质的功能丧失或获得有关 [11]。例如,复制蛋白 A 是一种参与 DNA 修复、复制和重组的蛋白质。乳腺癌 2 号基因(breast cancer 2,BRCA2)发生癌症易感性突变使其削弱其与复制蛋白 A 的相互作用 [12]。缺乏这种相互作用可能会抑制对 DNA 修复蛋白的招募,并最终导致致癌 DNA的积累。此外,肿瘤抑制蛋白 P53 在细胞受到内/外刺激(例如 DNA 损伤、癌基因活化或氧化应激)时被激活,通过启动细胞周期阻滞、细胞衰老、DNA 修复和细胞凋亡等途径抑制肿瘤细胞的产生 [13]。反之,P53 突变会破坏复杂的应激反应途径网络,导致受损细胞失控增殖,并最终导致各种类型的肿瘤发生 [14]。因此,PPI 已成为生物化学和药物化学中重要且具有挑战性的目标 [15]。PPI 调节剂的开发将有助于提高对 PPI 网络的认识,促进对疾病致病机制的理解,从而为开发新的诊断治疗策略铺平道路 [9]。多肽由于其独特的优势,在 PPI 调节药物开发方面具有极大的应用潜力。迄今为止,已经报道大量成功的多肽类 PPI 调节剂,其中在克服多肽成药瓶颈中付出的努力令人印象深刻。对此,本文综述近几年靶向 PPI 多肽在抗肿瘤药物中的应用、多肽药物设计方法和基于纳米工程化的多肽递送手段等方面的研究进展。
1靶向胞内蛋白-蛋白相互作用的多肽药物抗 肿瘤机制
相比于小分子药物,多肽类药物具有庞大的表面积,与靶蛋白的结合界面更大 [16]。因此多肽类PPI 调节剂对靶点具有更高的亲和力和专一性,可以发挥更大的药效,同时有效降低相关治疗副作用。此外,由于多肽抑制剂表面上具有多个相互作用位点,个别结合位点的缺失不会导致结合能力的完全丧失,这一特点使其对靶蛋白结构的细微变化具有更高的容忍性。选择合理的治疗靶点是肿瘤靶向治疗的关键问题之一,直接决定着肿瘤靶向治疗的成败,针对不同靶点的 PPI 其抗肿瘤机制也可能不同。本文将靶向 PPI 多肽药物的抗肿瘤机制分为以下 3种:直接靶向 PPI 抑制肿瘤细胞、抑制肿瘤免疫相关 PPI 靶点增敏肿瘤免疫治疗以及调控肿瘤微环境相关 PPI 增敏肿瘤治疗。下面将对近年来针对肿瘤治疗的重要靶点进行讨论。
1.1 直接靶向 PPI 抑制肿瘤细胞
肿瘤的发生和发展通常与细胞中某些蛋白的异常表达与降解有关。通过多肽药物的设计直接靶向抑制或激活肿瘤相关靶点蛋白的表达,是靶向 PPI常见的抗肿瘤机制。下面对此类抗肿瘤靶点及相关的多肽药物进行详细叙述。
1.1.1 P53 激活肽及其衍生多肽 P53 蛋白作为一种广为人知的肿瘤抑制因子,可响应 DNA 损伤或癌基因激活等细胞应激而调节多种靶基因的表达,从而介导细胞周期停滞、DNA 修复、衰老或凋亡等细胞活动 [17]。据不完全统计数据,50% 以上的恶性肿瘤细胞中 P53 基因发生异常,P53 蛋白编码基因 TP53的变异或缺失在 12 种最常见的肿瘤类型中占比约为42%。突变的 P53 蛋白不能调节 DNA 损伤导致的细胞生长停滞和细胞死亡,直接导致肿瘤的恶性进展和不良预后。因此恢复内源性 P53 蛋白活性可以诱导细胞凋亡、衰老和先天性炎症反应来阻止体内恶性肿瘤的生长。
在许多肿瘤中,P53 基因以野生型形式存在。而野生型 P53 基因受到多种机制的负调控不能发挥正常功能。这些负调控机制的核心,是鼠双微体基因 2(murine doubleminute 2,MDM2)和鼠双微体基 因 X(murine doubleminute X,MDMX)对抑癌蛋白 P53 的协同抑制 [18-20]。因此,同时阻断转录蛋白 MDM2 和 MDMX 对 P53 蛋白的负调控,从而激活肿瘤细胞中的 P53 蛋白是治疗携带野生型 P53 基因癌症患者的一个合理思路。Pazgier 等 [21] 通过筛选随机十二肽噬菌体文库,鉴定几种 P53-MDM2/MDMX 抑制肽,其中最有效的多肽抑制剂 PMI(多肽 序 列:TSFAEYWNLLSP)以低浓度亲和力与MDM2 蛋白和 MDMX 蛋白结合,比相同长度的野生型 P53 肽(多肽序列:ETFSDLWKLLPE)结合力强约 2 个数量级。该发现破译 P53-MDM2/MDMX高亲和力抑制肽的结构基础,为不同类别 P53 激活剂的结构设计提供新的依据以及潜在的治疗用途。
1.1.2 β-连锁蛋白抑制肽及其衍生多肽 B 细胞淋巴瘤因子 9(b-cell lymphoma 9,Bcl9)是一种在人类肿瘤中过度表达的转录辅因子。Bcl9 与 β-连锁蛋白(β-catenin)相互作用激活 Wnt/β-catenin 信号,促进其核易位,从而促进肿瘤进展 [22]。尽管 Wnt/β-catenin 途径在肿瘤发生中起着关键作用,但它也是干细胞增殖和分化所必需的 [23],所以对该途径进行治疗性干预仍然是一个挑战。因此,靶向 Wnt/β-catenin 途径并对肿瘤细胞具有高度特异性的药物开发将具有重要的临床意义。
短肽 Bcl9 中的 3 个疏水残基和 2 个带电残基可与 β-catenin 中的疏水沟相互作用。因此,该螺旋线很有可能用来阻断 Wnt 信号的活性。为此笔者课题组合成了 β-catenin/Bcl9 抑制肽(β-catenin/Bcl9inhibitor,BBI)[24]。基于荧光偏振的竞争结合分析表明 BBI 与 Bcl9 显著竞争结合 β-catenin。通过与细胞穿透肽 R8(多肽序列:RRRRRRRR)相连接,结合肽 BBI-R8 对 Wnt 信号超活化细胞系 Hep3B 的增殖表现出强大的抑制能力。这些结果表明,多肽BBI 能通过阻断 Bcl9 和 β-catenin 的相互作用来抑制癌细胞的增殖。笔者课题组进一步设计 pH 和谷胱甘肽(glutathione,GSH)双响应的纳米杂合物团簇 [24]。一系列体外和体内测试表明,该纳米杂合物通过阻断 Wnt/β-catenin 通路,有力地抑制肿瘤生长和转移,同时保持了高度有利的生物安全性。以上结果表明多肽 BBI 可以有效抑制 Wnt 信号通路的超活化,具有巨大的临床应用潜力。
多肽自组装平台以其独特的生物相容性和生物降解性,在多靶点抗肿瘤药物领域具有巨大的发展潜力,受到越来越多的关注。通过表位嫁接和结构设计,笔者课题组将以下 2 种抗癌肽引入丝瓜种子蛋白(一种天然蛋白质)的拓扑等效位置:1)PMI,P53-MDM2/MDMX 十 二 肽 抑 制 剂;2)Bcl9p,一种通过阻断 β-catenin-Bcl9 相互作用来抑制 Wnt/β-catenin 信号的多肽。通过此多肽嫁接技术设计一种基于多肽的杂交纳米平台 Lupbin(见图1)[25]。它能够自组装成一个稳定的结构,并同时靶向位于细胞质和细胞核的多个 PPI。结果表明,Lupbin 通过同时阻断 Wnt/β-catenin 信号通路和激活P53 信号通路来阻断肿瘤的生长和转移,具有良好的体内生物安全性。
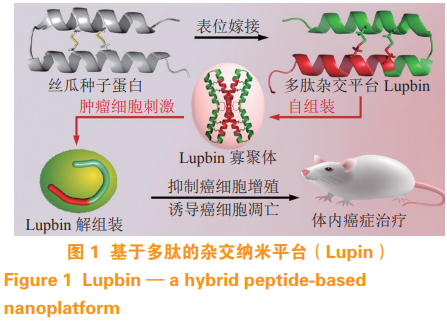
1.1.3 其他靶点 除上述靶点之外,最近研究发现了许多新的肿瘤治疗靶点。肿瘤相关成纤维细胞(cancer-associated fibroblast,CAF)广泛存在于多种肿瘤中,在肿瘤进展和免疫抑制中起着关键作用[26]。成纤维细胞激活蛋白(fibroblastactivationprotein,FAP)在 CAF 上过表达,被认为是一个普遍的肿瘤靶点。然而,FAP 的表达并不局限于肿瘤,针对 FAP 的全身治疗往往会导致严重的副作用 [27]。为此,Zhou 等 [28] 开发一种铁蛋白纳米颗粒介导的FAP 靶向光动力疗法。实验结果表明,该药物系统中 FAP 特异性单链可变片段可以有效地靶向并根除肿瘤中的 CAF。
近年来,肿瘤细胞内的细胞核和重要的细胞器如线粒体和内质网等成为许多癌症治疗策略的重要靶点 [29-31]。最新研究表明,线粒体在癌症的耐药性中起到关键作用。在此,He 等 [30] 利用线粒体周围酶的自组装选择性靶向肝癌细胞线粒体。包裹着氯霉素(chloramphenicol,CLRP)的肽-脂质结合物最初被线粒体周围肠激酶切割,随后 CLRP 被选择性地送入癌细胞线粒体,从而抑制线粒体蛋白质合成,诱导细胞色素 c 释放到胞浆中,最终导致癌细胞死亡。
1.2 通过抑制肿瘤免疫相关蛋白-蛋白相互作用靶点增敏肿瘤免疫治疗
人造血系统、免疫系统或炎症反应中的活化细胞可以产生具有高活性和多功能的肽、蛋白质和糖蛋白。它们在机体的免疫活化和免疫抑制进程中起着重要作用。肿瘤免疫疗法通过激活体内免疫细胞达到去除肿瘤细胞的目的。肿瘤免疫疗法具有特异性强、作用时间久和副作用少等优点 [32]。通过肽库筛选、序列比较或其他相关技术来获得可以调节肿瘤患者免疫力的多肽,将在肿瘤治疗中发挥积极作用 [33]。
程序性细胞死亡蛋白-1(programmed cell deathprotein 1,PD-1)是一种众所周知的免疫检查点受体,它在肿瘤组织的 T 细胞中过度表达。Han 等 [34] 报道一种靶向吲哚胺 2,3-双加氧酶(indoleamine-2,3-dioxygenase,IDO)的纳米抑制剂,其可以选择性地抑制肿瘤微环境中的 IDO 通路。该抑制剂是由亲水性靶向分子、2 个可质子化的组氨酸和 1 个连接有酯键的疏水性 IDO 抑制剂组成,其具有 pH 响应解聚和酯催化的药物释放性能。该抑制剂可以对肿瘤内 IDO 活性进行强力持久的抑制,从而极大地增强程序性细胞死亡配体 1(programmed cell deathligand 1,PD-L1)在体内的治疗效果。Li 等 [35] 报道 一 种 近 端 使 能 反 应(proximity-enabled reactivetherapeutics,PERx)的方法来产生共价蛋白药物。通过遗传密码扩展,潜在的生物反应性氨基酸氟硫酸 盐-L-酪 氨 酸(fluorosulfate-L-tyrosine,FSY) 被整合到 PD-1。当 PD-1 与 PD-L1 相互作用时,FSY才会选择性地与 PD-L1 的近端组氨酸反应,从而在体外和体内都能使 PD-1 与 PD-L1 不可逆地结合。在免疫人源化小鼠模型中,与非共价野生型 PD-1相比,共价 PD-1-FSY 表现出更强的抗肿瘤作用,并达到等效或优于抗 PD-L1 抗体的治疗功效。
肿瘤干细胞(cancer stem cell,CSC)是具有干细胞性质的肿瘤细胞,并能驱动肿瘤转移和复发。迄今为止,已经在 20 多种肿瘤中发现 CSC。Najafabadi等 [36] 从 CSC 中鉴定出乙醛脱氢酶(acetaldehydedehydrogenase,ALDH)的 A1 和 A3 表位,并研制了用于预防 ALDHhigh CSC 的合成高密度脂蛋白纳米盘。纳米盘增强了抗原向淋巴结的转运,并产生了强大的 ALDH 特异性 T 细胞反应。该纳米盘疫苗联合抗 PD-L1 治疗在多种小鼠模型上均显示出较强的抗肿瘤效果,并可能为 CSC 的癌症免疫治疗开辟新的途径。
1.3 调控肿瘤微环境相关蛋白-蛋白相互作用增敏肿瘤治疗
长期以来,肿瘤微环境(tumor microenvironment,TME)在肿瘤研究中一直被忽略。受 Paget“种子与土壤”假说的启发,越来越多关于 TME 的研究取得令人瞩目的成就 [37-39]。TME 由肿瘤部位的所有非恶性宿主细胞和非细胞成分组成,包括肿瘤相关成纤维细胞(tumor-associated fibroblasts,TAF)、细胞外基质(extracellular matrix,ECM)、肿瘤相关免疫细胞和内皮细胞。所有这些成分都引起强烈的促结缔组织增生反应,极大影响治疗剂的输送效率和治疗效果。因此,已经开发出许多靶向调节 TME相关 PPI 的多肽药物来阻止肿瘤的发展 [40]。如促进ECM 相关成分 ( 胶原蛋白和透明质酸等 ) 的降解,可以释放肿瘤位点增加的组织间渗透压,从而提高治疗剂的效率。
炎症反应,特别是慢性炎症,是引发肿瘤的重要原因,并在肿瘤的不同发展阶段发挥不同的功能。减少炎症的免疫调节以重塑 TME 可能是癌症治疗的重要策略。Ma 等 [41] 开发一种 pH 和氧化还原双重响应性多肽-地塞米松结合物(L-SS-DEX),用于抗肿瘤治疗和调节 TME。该药物不仅在体内和体外均对肿瘤细胞具有显著的抑制作用,而且显著增加 CD8+T 细胞的浸润,降低肿瘤内的髓系来源抑制性 细 胞(myeloid-derived suppressor cell,MDSC)和调节性 T 细胞的数量,成功逆转肿瘤的免疫抑制微环境。
此外,抑制肿瘤血管的形成和发展也是调节TME 治疗有希望的策略之一。鉴于此,Li 等 [42] 开发一种将凝血酶和化疗药物阿霉素(Dox)整合的共递送纳米系统。结果表明,该药物在肿瘤部位表现出凝血酶的受控释放,局部引发肿瘤内血栓形成,从而剥夺肿瘤的营养。同时 Dox 在肿瘤内的积累和扩散,对肿瘤细胞产生良好的杀伤效果。
2 靶向胞内蛋白-蛋白相互作用的多肽开发
多肽由于具有良好的生物相容性和结构多样性,已经成为靶向 PPI 调节最可能的候选药物。然而多肽本身的固有缺陷(蛋白水解抵抗性差和不良细胞内化等)限制治疗性肽 PPI 调节剂的开发 [43]。研究者为克服这些阻碍已经付出巨大的努力,并取得一系列有价值的成果。以下对多肽药物靶向 PPI的优势与障碍进行详细叙述,并对多肽药物的设计手段进行归纳总结。
2.1 抗肿瘤仿生多肽靶向蛋白-蛋白相互作用的优势与劣势
2.1.1 优势 PPI 的调节已经通过多种途径实现,特别是来自天然化合物或通过有机合成的小分子方法近年来被广泛研究 [44]。目前有超过 12 个小分子 PPI调节剂正在临床开发中,预计这些分子的市场价值将在未来 5 年内超过 6 亿欧元 [45]。尽管如此,小分子调节剂对绝大多数的 PPI 无明显效果。目前,制药行业开发特定 PPI 靶标小分子抑制剂的进展有如下困难:1)小分子药物参与调节 PPI 靶点的作用面积有限;2)小分子在结合蛋白相互作用界面中遇到强大的热力学障碍;3)难以在化学库中找到能够克服这些热力学障碍的合适化合物。实验证明克服这些障碍非常具有挑战性。通常对 PPI 起作用的小分子抑制剂,常规高通量筛选的命中率令人失望 [46]。
近年来生物药物(例如抗体、蛋白质和肽)正在作为诸如蛋白质相互作用的靶标抑制剂而不断出现。相比于小分子药物,多肽具有以下优势:1)肽本身具备柔韧性,对 PPI 大的相互作用表面有较强适应性;2)易于模块化,具有结构多样性,从而具有更高的选择性和效力;3)多肽的尺寸限制其在组织中的积累;4)良好的生物相容性 [47],如相比于小分子抑制剂 Nutlins,p52-MDM2/MDMX 靶向抑制肽 PMI 对 MDM2/MDMX 具有更高的亲和力 [21]。
2.1.2 劣势 尽管多肽与小分子相比具有一些优势(如对靶点的亲和力/特异性较高、毒性较低),但由于多肽的固有缺陷,其一直无法成为候选药物的主要来源。首先,多肽药物仅限于已知的生物活性肽靶点,并且表现出较低的效能。此外,由于人体内肾脏的快速清除和血液、肾脏或肝脏的酶消化,肽的短半衰期一直是肽疗法的主要问题之一。肽通常在给药后几分钟到几小时内从血液中清除。因此,目标组织可能缺乏足够的暴露,使药物难以产生体内效应。此外,多肽药物开发也受到较低的细胞膜通透性和可能包含潜在的免疫原性序列的限制 [48]。
针对这些药理障碍已经开发许多策略,如通过聚乙二醇(polyethylene glycol,PEG)对目标肽进行修饰还可以保护其免受外来的影响,从而提高其在体内的整体稳定性。另外,PEG 修饰可以显著降低某些肽(非普遍)的免疫原性 [49]。利用血清白蛋白可以逃避肾脏清除的特性,将干扰素 α 肽融合到白蛋白 C 端,在保障药物效力的同时可以有效延长药物半衰期。将肽共价连接到血清白蛋白氨基酸 34 位的游离半胱氨酸也能有效延长治疗肽的半衰期 [50]。近年来,纳米技术提供一种自下而上的方法,可将肽共价或非共价地转化为具有蛋白水解抗性和细胞膜穿透性的稳定结构。一些肽衍生的纳米药物,包括脂质体及大分子衍生的肽纳米胶束,肽涂覆的纳米颗粒和基于肽的自组装纳米结构表现出优越的生物学优势,包括延长体内循环时间、增强疾病特异性、增强蛋白水解稳定性和随后优化的治疗功效 [51]。
2.2 靶向蛋白-蛋白相互作用的多肽药物开发手段
2.2.1 结构辅助设计 许多PPI是由小段多肽介导的,它们具有特定的二级结构以适应相互作用的表面。然而蛋白质片段作为 PPI 调节剂存在一些局限性,特别是当从整体蛋白质中分离出短序列多肽段时,往往会丧失其高度的灵活性和三维稳定性。对蛋白质片段或多肽进行结构辅助设计以增强其作用效果,是多肽药物开发的常用手段 [46],例如提高药物蛋白水解抗性、靶点亲和力以及增强细胞内化等。
Schafmeister 等 [52] 于 2000 年报道通过复分解反应可以诱导短肽中的 α-螺旋构象。α-螺旋肽由于具有更好的亲和力、更高的稳定性和良好的细胞摄取能力,在调节 PPI 的多肽药物开发中得到广泛应用。PPI 成功被调节的例子有原癌基因 B 淋巴细胞瘤-2 基因(B-cell lymphoma-2,BCL-2)转录产物抗凋亡蛋白 BCL-2 家族、抗凋亡基因髓样细胞白血病-1 基 因(myeloid cell leukemia-1,MCL-1)转录产物 MCL-1 蛋白 [53]、P53-MDM2 相互作用 [54-55]、Notch 信号通路中形成的信号分子三元复合物 [56] 和Wnt 信号通路中的 β-catenin/T 细胞因子 [57]。订书肽被用来抑制新的抗癌靶点,如通过阻止白血病细胞中 Zeste 同源物增强子 2 重组蛋白(enhancer of zestehomolog 2,EZH2)与胚胎外胚层发育蛋白(embryonicectoderm development,EED) 复 合, 并 通 过 降 低EZH2 蛋白水平来抑制组蛋白 H3 第 27 个氨基酸的三甲基化 [58]。
肽介导的蛋白质相互作用还涉及另一种弥漫的二级结构,即 β-发夹结构。这种结构通常由 2 条反平行的 β-链组成,并由 1 个环序列连接。许多策略都致力于提高这种发夹的稳定性,以调节 PPI [59]。β-发夹稳定肽还可以模拟 P53 蛋白 N 端片段中的 α螺旋表位,并以纳摩尔级亲和力与 MDM2 结合 [60]。受抗体结构的启发,稳定的 β-发夹结构被用于构建一类新的高效结合肽,该结合肽对多个靶点显示出纳摩尔级亲和力和较慢的解离速度。
2.2.2 计算机辅助蛋白设计 计算机辅助蛋白设计(computer aided protein design,CAPD)是指应用计算机技术对蛋白质的结构预测和改造方案进行评估,并作出最佳选择。目前常见的蛋白质设计软件主要有 PyMOL,PDB Viewer,Accelery Insight II 和Discovery Studio 等软件。相较于人力,计算机辅助设计具有容量大、速度快和分析能力强等诸多优势。尤其是随着计算机硬件和算法的快速升级,其分析能力和速度得到极大的提升。因此计算机辅助设计具有极大的发展潜力和应用前景。Senior 等 [61] 描述一种 AlphaFold 算法,通过引入机器学习技术,极大提高蛋白质结构预测的速度和精度。
此外,应用计算机技术还可以对改造的蛋白质进行预测。Sun 等 [62] 基于成纤维细胞生长因子 2(fibroblast growth factor 2,FGF2)与成纤维细胞生 长 因 子 受 体(fibroblast growth factor receptor,FGFR)形成的复合物结构,制备 3 种 FGF2 的变体,并基于计算机辅助设计揭示了蛋白质的结构特征。结果表明,对蛋白质进行有效的修饰可以提高其活性和稳定性。这种蛋白质修饰策略(如结构导向的聚乙二醇化)为蛋白质改造提供了新的方法。
2.2.3 多肽库筛选 1)噬菌体展示 将生物技术用于药物探索已经成为现实。其中噬菌体展示是靶向PPI 多肽药物开发的常用工具,并广泛用于为高通量筛选(high throughput screening,HTS)建立各种肽库 [46]。众所周知,该方法具有高变异率以及“亲和力选择”的独特优势 [63]。如 Zhou 等 [64] 发现一个新的免疫检查点:含免疫球蛋白和 ITIM 结构域蛋白的 T 细胞免疫受体(T cell immunoreceptor with Igand ITIM domains,TIGIT),并使用镜像噬菌体展示对 TIGIT 的 D-对映体进行生物淘洗。筛选出的D 肽 D-TBP-3 可以占据 PPI 结合界面,并有效阻断TIGIT 与脊髓灰质炎病毒受体(poliovirus receptor,PVR)的相互作用。尤其在抗 PD-1 耐药肿瘤模型中,D-TBP-3 可以抑制肿瘤的生长和转移,有望成为肿瘤免疫治疗的潜在候选药物。
Li 等 [65] 对噬菌体进行基因工程改造,使其侧壁上显示出许多已鉴定的血管生成素结合肽副本,并在其尖端上显示出多个乳腺肿瘤归巢肽副本。由于可以通过噬菌体展示发现归巢肽,并且可以针对任何特定癌症进行定制,这种血管生成素结合噬菌体是通用的“即插即用”肿瘤归巢肽治疗药物。Kong 等 [66] 基于噬菌体的进化和选择开发一种生成目标特异性肽(相对分子质量小于 1 600)的方法。通过该方法产生具有纳摩尔级亲和力的凝血因子抑制剂,并为白介素 23 受体开发了一种抗肠胃蛋白酶的肽拮抗剂。该方法可用于设计胃肠道稳定且亲和力高的靶向肽,有助于口服多肽药物的开发。
2)核糖体和 mRNA 展示 体外技术的发展已解决在活细胞中建立随机肽库的局限性,如核糖体展示和 mRNA 展示等,这些技术不仅与噬菌体展示同样具有高度多样性,并且效率得到显著提高 [67]。
在核糖体上展示的随机肽库正在成为体外选择生物学相关大分子(包括表位、拮抗剂、酶和细胞表面受体)的新工具。核糖体展示是一种无细胞的系统,通过形成稳定的蛋白质-核糖体-mRNA复合物,并通过淘选和反向的迭代循环选择功能性的新型蛋白质,从而将各个新型蛋白质(表型)与其相应的mRNA(基因型)偶联 [68]。基于此方法,Yang 等 [68]使用大肠埃希菌 S30 裂解物构建了高复杂度的随机肽库(肽库中多肽数目超过 1014 个)。此外,核糖体展示已用于筛选雌激素受体表面,并发现了新的富含脯氨酸的肽结合剂 [69]。
通过生物技术制备肽库的最重要进展之一是遗传密码的合成重编程,它允许在肽序列中同时引入多个非蛋白质氨基酸。该技术涉及将 mRNA 展示与突变的氨酰 t-RNA 合成酶结合使用,该酶可催化包含给定氨基酸反密码子序列的 RNA 链的氨酰化 [70]。Morimoto 等 [70] 开发了更多用途的氨酰化催化剂,并与适当的翻译系统配合使用,使该系统完成蛋白质和含非天然氨基酸肽的合成工作 [71]。使用 mRNA展示的实例是成功分离与凝血酶低亲和力的非天然环状肽 [72],以及发现拮抗活细胞中血管内皮细胞生长因子受体 2(vascular endothelial growth factorreceptor 2,VEGFR2)的大环状肽 [73]。
3 靶向胞内蛋白-蛋白相互作用的多肽成药性改造
多肽具有以下缺陷:1)抵抗消化系统蛋白水解酶并且血浆降解的稳定性低;2)容易被肝脏和脾脏快速清除;3)穿越生理障碍的能力差;4)潜在的免疫原性等,极大地限制其作为 PPI 调节药物的开发。为此,许多化学修饰和智能连接体已被引入PPI 调节剂的开发 [74]。这些化学修饰不仅使多肽药物具有细胞和组织通透特性,还可以降低蛋白质水解降解和提高生物利用度。
近年来,抗肿瘤纳米系统作为药物载体得到广泛应用。由于肽的生理功能的多样性,将肽引入抗肿瘤纳米系统是合理的。肽与纳米材料的集成具有互补优势,不仅避免体内肽的快速降解,而且使纳米系统更加智能化和功能多样化 [75-76]。下面对多肽药物中常见的化学修饰,以及基于抗肿瘤纳米系统的多肽递送手段进行介绍。
3.1 基于多肽修饰的化学手段
化学修饰是设计多肽的重要手段。其中,侧链结合和主链环化等化学修饰方法被广泛应用。
3.1.1 主链环化 环肽可分为 2 种类型:均环肽和杂环肽。均环肽本质上是形成分子内肽键的过程。影响环肽形成的因素是环的大小,如具有 7 个以上氨基酸残基的多肽可以顺利成环。存在于杂环肽的氨基酸残基之间的非酰胺键主要是酯键和二硫键。前者与酰胺键的形成过程相似,而后者实质是将 2 个半胱氨酸的巯基氧化成二硫键。
较短的线性肽很容易被生物体内的各种生物酶降解。由于环状肽不具有 C 末端和 N 末端,因此可以消除或减弱肽酶的降解作用,从而提高多肽在体内的稳定性。Wang 等 [77] 报道一种超分子管水凝胶(tubustecan,TT),通过组织穿透环肽 iRGD 与2 个喜树碱药物单元的共价连接产生一种 TT 前药两亲体,可结合成管状超分子聚合物。其中 iRGD序列既是肿瘤结合肽又是组织穿透肽。完整的环状肽可与肿瘤相关内皮细胞表面过度表达的 αv 整合素结合。iRGD 切割后的 RGDR 序列可以通过与肿瘤细胞过度表达的血管内皮生长因子(vascularendothelial growth factor,VEGF)和神经纤毛蛋白 1(neuropilin-l,NRP-1)结合来刺激药物在肿瘤中的穿透和促进细胞内吞。结果表明,该药物载体有长期持续的药物释放时间。由于 iRGD 环肽的整合,药物有效地渗透到实体肿瘤,并得到增强的治疗效果。
同时由于环状结构对构象的限制增加,有可能提高肽与受体的亲和力和选择性,从而增强多肽药物活性并降低副作用。Shen 等 [78] 进行的研究表明,对于一系列实体瘤,用近红外染料标记的环状八肽可选择性结合磷酸化膜联蛋白 A2(annexin A2,ANXA2),且在高钙水平下具有高亲和力。该发现表明,大多数实体瘤的微环境中都存在高水平的ANXA2 磷酸化与钙含量升高。八肽可能广泛用于选择性肿瘤成像以及作为载体将药物递送至实体瘤部位。α-促黑素细胞激素环状类似肽可以有效靶向肿瘤细胞表面过度表达的黑皮质素受体。Zhang 等 [79]在二氧化硅纳米颗粒上修饰 α-促黑素细胞激素环状类似肽,以提高对肿瘤细胞靶向能力。结果表明,该纳米颗粒在同基因小鼠黑色素瘤细胞和人黑色素瘤细胞移植模型中具有选择性的肿瘤摄取和良好的生物分布特性。
3.1.2 侧链修饰 非标准氨基酸的加入不仅可以改善肽的理化性质,而且可以增加肽的新功能 [80]。多肽的组装后修饰是获得功能化多肽并用于生物学研究和药物开发的有效途径。大多数肽的修饰方法主要是基于氨基、羟基、羧基和硫醇等“活性”官能团的转化 [81],对酪氨酸、色氨酸、苯丙氨酸和组氨酸的芳香 C(sp2)-H 功能化也进行广泛的研究。相比之下,由于肽链的脂族侧链的惰性和控制区域选择性的困难,对 C(sp3)-H 功能化的研究报道较少 [82-83]。
近年来,已开发几种用于 N- 或 C- 末端肽的C(sp3)-H 功能化的定向策略。Rodríguez 等 [84] 报道通过在 N- 末端安装双齿辅助剂来实现 N- 末端残基Val、Ile 和 Tle 的 γ/δ-C(sp3)-H 功 能 化, 而 Mondal等 [85] 利用 C- 连接的双齿辅助剂作为导向基团开发了 C- 末端 Pro 和 Ala 的 β-C(sp3)-H 芳基化。在没有辅助剂的情况下,N- 端和 C- 端的天然氨基和羧基也被用作导向基团,其分别促进短肽的 N- 端或 C端 Tle 的 γ-C(sp3)-H 芳基化。此外,Weng 等 [86] 报道了由天冬酰胺的侧链酰胺基促进的 N- 末端 Ala的 β-C(sp3)-H 芳 基 化。此 外,Gong 等 [87] 开发了一种主链导向策略,并实现了 N- 末端 Ala 的高效β-C(sp3)-H 芳基化、乙酰氧基化和炔基化。使用该策略,Tang 等 [88] 进一步报道利用线性肽的分子内β-C(sp3)-H 芳基化合成环肽。同时 Mondal 等 [85] 也报道了 β-C(sp3)-H 功能化三唑类肽,并且这些方法被成功地应用于修饰内部 Ala。
3.2 基于纳米工程化的递送策略
目前,已经设计和利用各种纳米粒子来增强多肽物理和生物稳定性,以促进细胞和组织渗透,并有效地促进细胞内多肽递送。包括脂质体、聚合物、蛋白质复合物、无机材料在内的各种纳米载体系统已被证明是有希望的递送策略 [51]。
3.2.1 基于囊泡的递送策略 1964 年,Bangham 等 [89]发现脂质体可以作为输送囊泡,随后这种载药系统得到广泛研究。截至目前,已经有几种基于小分子药物的脂质体产品被美国 FDA 批准 [90]。脂质体是具有内腔的双层囊泡,由两亲性脂质或磷脂组成。由于其特殊的结构,可以将各种疏水性或亲水性药物填充到脂质体中,然后通过内吞作用或脂质体细胞融合作用传递到细胞中。例如,为提高治疗效果,Jiang 等 [91] 设计凝胶-脂质体复合物共传递 2 种抗癌药物,即肿瘤坏死因子相关的凋亡诱导配体(TNF related apoptosis inducing ligand,TRAIL)和 Dox。在此,Dox 首先被封装到由细胞膜穿透肽修饰的脂质体中。由酶降解的透明质酸(hualuronicacid,HA)组成的交联凝胶附着在脂质体的表面。同时,抗癌剂 TRAIL 被包封在外壳中。透明质酸酶在 TME 中高度过表达,这可能会降解 HA 外壳,从而促进 TRAIL 与死亡受体结合并促进肿瘤细胞对Dox 的摄取。体内试验表明,这种多功能凝胶-脂质体系统对人乳腺癌细胞(MDA-MB-231)异种移植动物模型的肿瘤生长有明显的抑制作用。
近年来,外泌体作为一种药物递送系统受到越来越多的关注 [90]。作为一种内源性囊泡,外泌体具有低免疫原性、低毒性和较长的血液循环时间等优势。Li 等 [92] 开发了一种巨噬细胞外泌体包被的纳米平台,并将其用于三阴性乳腺癌的靶向药物递送和化疗。在外泌体膜表面修饰可以特异结合肿瘤细胞上过表达的间质-上皮转化因子的多肽,以提高对肿瘤的靶向效果。实验结果表明,该外泌体膜包被的纳米颗粒可以显著提高 Dox 的细胞摄取效率和抗肿瘤效果,并且该纳米载体在体内具有良好的肿瘤靶向效应和抑制效果。
3.2.2 基于仿生纳米颗粒的递送策略 在不同的仿生策略中,利用细胞膜材料制备纳米颗粒提供了一种独特的自上而下的方法,该方法具有能够完全复制源细胞表面抗原多样性的优势,并且避开了蛋白质组学方面的研究和多价纳米颗粒功能化的工程障碍。使用这种新兴方法,研究人员成功地创建了具有许多理想功能的纳米颗粒。包括具有长循环特性的红细胞膜覆盖的纳米颗粒,具有癌症靶向能力的干细胞膜来源的纳米颗粒,和具有穿越内皮细胞能力的白细胞膜涂层的纳米颗粒。癌细胞膜覆盖的纳米粒子具备同源靶向能力和免疫逃逸能力,被广泛用于抗肿瘤纳米颗粒的设计 [93]。Fang 等 [94] 报道一种基于 FePSe3 的仿生 2D 纳米片,其中抗 PD-1 肽作为纳米片的内部成分,而结肠癌 CT26 细胞膜作为纳米外壳,形成具备核磁共振成像和光声成像能力以及协同光热和免疫疗法的抗癌剂。试验结果表明,由于癌细胞膜的覆盖,纳米颗粒具有良好的同源肿瘤细胞内化能力和免疫逃逸能力。此外,抗 PD-1肽可阻断 PD-1/PD-L1 通路并激活细胞毒性 T 细胞,从而产生强大的抗肿瘤免疫效果。
在自然界中,许多病原体,例如病毒和细菌,在纳米级或微米级都具有柔软的棒状几何形状,它们具有高度渗透能力 [95]。受这一现象的启发,有研究报道与病原体类似的纳米颗粒具有很强的肿瘤富集能力和细胞内在化能力。为此,笔者课题组通过诱导硫醇自组装构建一种柔性镧系元素-肽纳米棒(lanthanide-peptide-derived nanorod,LProd)[96]。作为肿瘤诊断和肽衍生疗法的治疗平台,LProd 具有生物相容性好、稳定性强和荧光强度高的特点。笔者课题组将 P53-MDM2/MDMX 抑制剂 PMI(多肽序列:TSFAEYWNLLSP)引入 LProd。为了增强肿瘤靶向,在 LProd 表面组装了白细胞分化抗原 13(CD13) 特异性结合环肽 iNGR(多肽序列:CRNGRGPDC)。实验表明,LProd 具有比球形镧系元素 – 肽纳米颗粒更强的肿瘤特异性积累,并通过 PMI 激活肿瘤抑制蛋白 P53,有力地抑制人结肠癌小鼠模型中的肿瘤生长。
3.2.3 基于无机/有机颗粒的递送策略 无机纳米粒子具有尺寸可调、表面改性等优点,能有效地进入靶细胞,且毒性小、循环时间长,是近年来研究的热点。无论是通过非共价相互作用还是共价连接,多肽药物都可以被这些纳米粒子包裹。目前广泛使用的无机纳米颗粒主要有硅纳米颗粒、金属纳米颗粒(aurum nanoparticles,AuNP)、碳基纳米材料等。硅基纳米颗粒具有成本低、生物相容性好、理化稳定性好、表面功能化程度高、临床发展水平高等特点,已被广泛应用于药物递送的载体 [97]。如 Nie等 [98] 开发了一种以介孔二氧化硅纳米粒子为支撑的蛋黄壳结构的纳米颗粒载体。结果表明,该封装有小剂量 Dox 和聚(ADP-核糖)聚合酶抑制剂的纳米载体比一线化疗药物盐酸阿霉素有更显著的抗肿瘤作用。
金属基纳米颗粒由于其尺寸小、形状不可变和种类多样性等优点被认为是一种很有前途的给药系统。其中以 AuNP 为基础的纳米载体具有良好的理化稳定性和生物相容性,因此,基于 AuNP 的疗法已被广泛应用于临床试验中。然而,肽的复杂化学性质(疏水性、电荷和氧化还原)始终不利于结合后胶态 AuNp 的稳态,随后导致在离子浓度升高的生理条件下的聚集甚至沉淀 [51]。对此,笔者课题组将 HAuCl4 和 β-catenin-Bcl9 相互作用的抑制肽 BBI共聚,形成金-肽纳米杂化物 [24]。这种纳米杂化物对多肽药物有较高的装载效率,而且具备低 pH 和GSH 响应特性。通过体外和体内测试,证明该纳米颗粒充分发挥抑制肽 BBI 对 Wnt/β-catenin 通路的损伤作用,有力地抑制肿瘤生长和转移,同时保持高度有利的生物安全性(见图 2)。
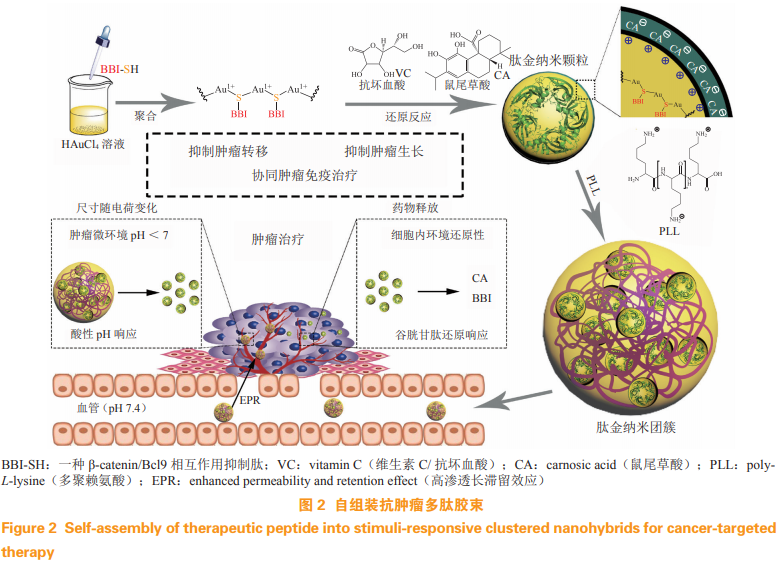
与聚合物、脂质、无机纳米颗粒等运输载体相比,蛋白质纳米粒子具有生物相容性、生物功能性、生物环境中的分子识别性、分子多样性和可生物降解成氨基酸等优点。目前,蛋白质载体已用于特定细胞靶向的药物递送。受丝瓜种子中的一种天然蛋白质的启发,笔者课题组设计了一种基于多肽的杂交纳米平台 [25],它能够自我组装成一个稳定的上部结构,并同时瞄准位于细胞质和细胞核的多个 PPI。结果表明,该纳米杂交平台能有效地穿透细胞膜,摆脱内质体依赖性降解,进而分解成游离单体,广泛分布于胞浆和细胞核中。重要的是,这种纳米平台可以发挥多肽药物的生物功能,且具有良好的体内生物安全性。笔者课题组的策略扩展自组装纳米药物在靶向细胞间 PPI 的应用,并可能为治疗包括肿瘤在内的各种人类疾病的肽治疗方法的发展注入新的活力。
此外,笔者课题组还开发了一种新的纳米工程大分子药物的新策略,即通过一种多肽 PSP(多肽序列:VVVVVHHRGDC)直接与药物结合成为PSP 药物单体,最终趋向于自组装成形状和电荷量由 TME 触发的纳米壳 [99]。作为概念证明,PSP 与肿瘤抑制因子 P53 的 D 肽激活剂 D-PMI(相对分子质量约为 1 492)结合生成直径为 80 nm 的空心球PSP-DPMI。实验结果表明,基于 PSP 的自组装成功地赋予 D-PMI 延长的体内循环时间和高肿瘤细胞特异性的细胞内积累。最终 PSP-DPMI 纳米壳通过激活 P53 信号通路在体内(外)有效地抑制肿瘤生长,同时保持良好的体内安全性。
4 结语与展望
尽管传统的肿瘤诊断和治疗策略在临床应用中具有许多优势(如良好的肿瘤抑制作用、众多的治疗评价和丰富的临床经验),但同时也面临着靶向性不理想、药物生物利用度低、副作用严重等诸多挑战。近几十年来,由于各种生物活性肽的独特优势,组合肽库的迅速发展成为推动针对肿瘤治疗的靶向PPI 多肽药物开发的强大动力。作为诸多抗肿瘤系统的组成部分,多肽药物可以靶向肿瘤中的 PPI 并对其进行调节。其独特的结构特点和特殊的生物活性,大大提高了治疗效果。目前,靶向 PPI 多肽药物有以下发展趋势:
1)治疗靶点的选择方面。大多数传统药物设计策略旨在设计针对一个靶点的具有高度选择性的化合物,以达到治疗效果。多肽由于其结构优势,可同时作用于多个治疗靶点。从治疗目标的疾病途径和 PPI 网络的角度出发,使多肽药物在 PPI 网络中表现出特殊的拓扑结构,从而达到更好的治疗效果。
2)提高多肽药物的稳定性和生物可利用性方面。设计具有理想的生物活性、稳定性和可生物利用性的多肽药物仍然是一项极其艰巨的任务。解决诸如蛋白降解和不良细胞内在化等问题是目前研究的主要目标。目前研究人员已经提出诸如肽修饰(如骨架循环、订书肽和非天然氨基酸掺入)等许多解决方案。尽管还有许多问题亟需解决,该领域仍有着巨大的发展潜力。
3)多肽药物的成药性改造方面。近年来纳米材料在抗肿瘤药物开发中具有独特的功能和各自的优势。靶向 PPI 多肽与各种纳米系统结合展示出前所未有的优越性和多功能性。多肽可以很容易地被整合到不同的纳米材料中,从而开发出具有优良肿瘤治疗效果的抗肿瘤靶向给药系统。
总之,靶向 PPI 多肽在肿瘤治疗中具有巨大的潜力,相信其在生物医学领域将发挥更大的作用。
参考文献



推荐阅读
Oyster Point Pharma宣布与箕星药业达成独家许可协议,在大中华区开发和商业化治疗干眼病的两款药物 诺华CEO入职3年学到的4件事:我在家开会,也得停下来去给儿子辅导作业;第一次上台紧张极了 辉瑞墨尔本工厂的脂质体和纳米颗粒技术介绍 诺华CEO畅谈改变他观念的五件事 头对头|被安进25亿美元收购的Teneobio和被明显低估的和铂医药 同类首创: 基石药业艾伏尼布在中国复发或难治性急性髓系白血病患者的注册研究达预期,新药上市申请已获中国国家药品监督管理局受理 感染8亿中国人的1类致癌物,不仅致癌,还会抑制免疫治疗效果!《BMJ》子刊最新研究 美国CDC发出警告:“德尔塔”的传染性与水痘相当,可以1传9…(附:PPT) 九月·上海|您不能错过的CMC峰会 罕见靶点不罕见,中国首款MET抑制剂获批背后,是和黄十余年的积累——「专访」和黄医药苏慰国博士 勃林格殷格翰2021创新大赛火热启动! 恒瑞VS百济:两种模式,殊途同归 又撤回一个?!PD-1你怎么了? 中国好BD|和黄医药
 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 