


⬆️欢迎参加2020中国NASH大会!
本文授权转载自药学进展,点击访问原文链接
专家介绍:杜涛

医学博士,美国汉佛莱医药顾问有限公司总经理,《药学进展》编委。杜博士在加拿大麦吉尔大学获得博士学位之后,师从美国科学院院士Frank Austen,在美国哈佛大学病理系从事了2年的博士后工作。在美国FDA担任高级审评官员7年,共审评过100多个药品的IND和NDA。在FDA工作期间,杜博士参与了FDA植物药指导文件和保健食品指导文件的起草,并为此而获得过FDA奖励。杜博士于2000年离开FDA后,曾先后在美国联合健保公司及和记黄埔医药任职。随后于2005年进入顾问行业,曾为多家欧美和中国的大型制药企业担任过医药开发顾问。杜博士也是北美金融界知名的技术顾问。他曾在3家北美的上市公司担任独立董事,并参与了多家公司的融资和上市。
医疗应对产品的相关立法

医疗应对产品激励政策
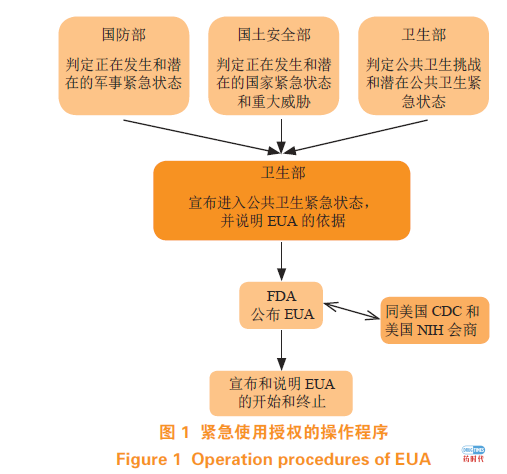
医疗应对产品紧急授权使用

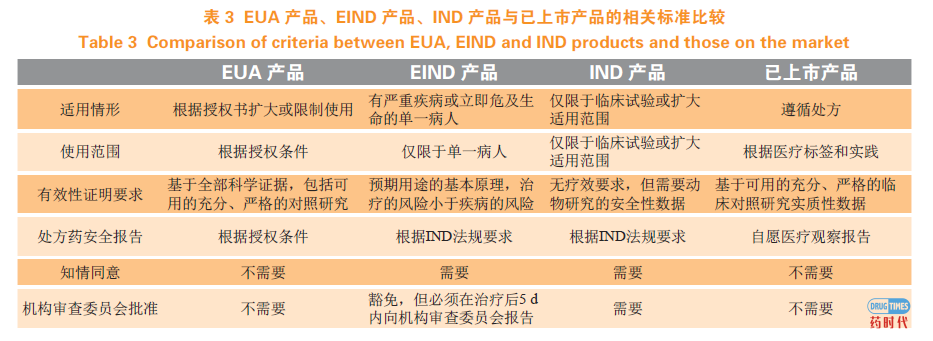
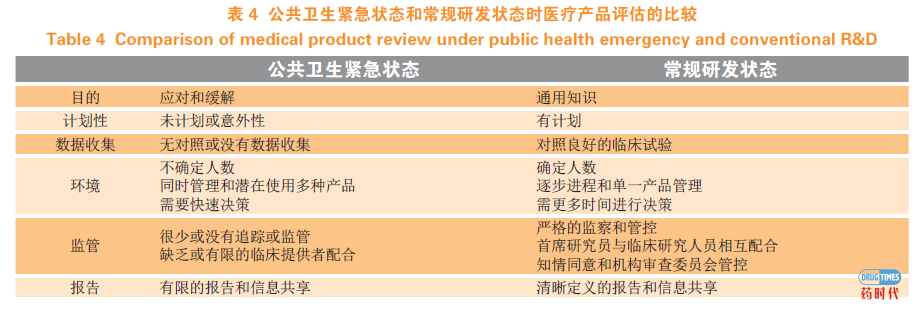
医疗应对产品的安全性和有效性评估在
医疗应对产品供应保障
FDA对公共卫生紧急事件的防范和反应

结语
版权声明:文中图片取自网络,根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系。衷心感谢!
研究进展:PD-1疗法响应低?匹兹堡大学针对LAG-3的新研究伸出援手
联盟喜讯|73家医院180余位专家学者参与!1类新药PDVⅢ期临床试验全国研究者会顺利召开!
瞬息万变的新冠疫苗赛道,Moderna刚公布数据就冒风险生产?
CRISPR先驱Jennifer Doudna发现超小型CRISPR酶—CasΦ(Cas12j),这是否将引领另一场技术革新?


 点击这里,与每月3万多朋友们欢聚!
点击这里,与每月3万多朋友们欢聚!
本文转载自药学进展,本文观点不代表药时代DrugTimes立场。
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 
