
正文共:1386字
预计阅读时间:6分钟
尽管一项II期临床试验未能达到主要终点(PFS),Imvax仍计划基于具有临床意义的总生存期(OS)向美国FDA提交IGV-001的生物制品许可申请(BLA)。
Imvax的底气何在?或许正源于其产品机制的创新性、以及显著的生存获益OS信号与当前监管科学趋势的高度契合。
创新性药械组合,与“死神”赛跑
IGV-001是一款针对新诊断多形性胶质母细胞瘤(GBM)的个体化肿瘤细胞疫苗,采用“生物制剂+医疗器械”组合模式,由三部分构成:患者的肿瘤细胞、IGF-1R反义寡核苷酸(IMV-001)、渗透室(其允许大分子渗出)。
其作用流程高度个体化:从患者手术切除的肿瘤组织中提取自体肿瘤细胞,经IGF-1R靶向反义寡核苷酸(IMV-001)处理,诱导受控细胞死亡;随后将这些处理后的细胞与过量IMV-001(作为免疫佐剂)共同装载于可植入的渗透室中,并在术后24小时内植入患者腹腔。48小时后取出装置,完成单次疫苗接种。
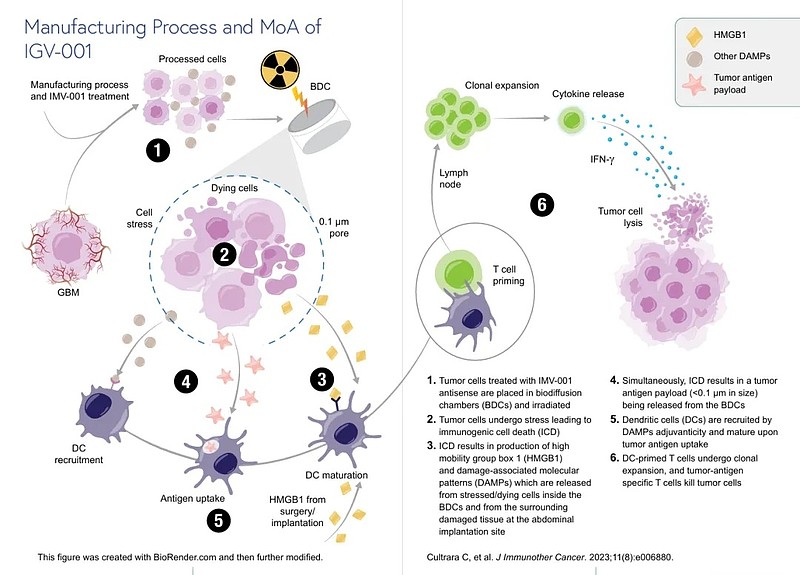

这一设计直面GBM治疗的核心挑战:血脑屏障(BBB)。Imvax指出,GBM患者在接受肿瘤切除术后,血脑屏障通常在一周内保持开放状态,而IGV-001将在GBM患者外科手术后的24小时内植入到患者体内,这为外周免疫系统识别肿瘤抗原并启动适应性免疫应答创造了关键窗口。
相比之下,标准治疗(包括放疗联合替莫唑胺化疗)通常需等待术后6周方可启动。而GBM生长迅猛、预后极差(中位生存期不足15个月),对患者而言,“时间就是生命”。IGV-001通过在术后黄金窗口期内激活全身抗肿瘤免疫,有望更早清除残留病灶,与“死神赛跑”。
OS展现显著获益,紧跟FDA当下的监管政策
再来看临床数据。该随机、双盲、安慰剂对照的IIb期试验共纳入99例新诊断GBM患者,在标准治疗基础上加用IGV-001或安慰剂。
尽管在预设的主要终点——无进展生存期(PFS)上,两组未显示出统计学显著差异,试验宣告未达主要目标,但关键次要终点OS展现出令人鼓舞的结果:IGV-001组中位OS达20.3个月,显著优于安慰剂组的14.0个月,相当于延长生存时间6.3个月(提升约45%)。

对于中位生存期通常不足15个月的GBM患者而言,“活得更久”是核心治疗诉求,而OS正是反映真实临床获益的“金标准”。
2025年,FDA肿瘤卓越中心发布《肿瘤临床试验中OS评估方法指南草案》,首次系统规范OS在随机对照试验中的统计设计与监管考量。该草案明确指出:无论OS是否作为主要终点,申办方均须在试验设计阶段预先制定完整的OS数据收集与分析计划,并将其作为支持药物疗效评价的核心依据。与此同时,CBER主任Vinay Prasad博士的回归进一步强化了这一趋势——他长期公开批评过度依赖PFS等替代终点,强调“以患者真实生存获益为中心”的审评理念,认为加速批准若缺乏OS支持,可能以患者治疗的不确定性为代价。
近年来,已有不少案例显示,即使临床试验未达到主要终点,具有突出临床价值或填补治疗空白的疗法仍可能获得FDA批准。其中最为著名的便是全球首款杜氏肌营养不良症(DMD)基因疗法Elevidys而闻名的Sarepta。
而放眼“同行”中,这样的案例仍历历在目。IO Biotech在其III期黑色素瘤试验中虽未达到PFS主要终点,但因观察到OS改善趋势,首席执行官 Mai-Britt Zocca 在投资者电话会议上表示,仍计划于2025年底前提交Cylembio的BLA申请。
结语
当然,BLA提交不等于获批。最终决定取决于FDA对整体风险获益的综合评估,以及Imvax能否提供更充分的临床证据。一款高度个体化的创新疗法能走多远,不仅需要监管认可,更需疗效验证与市场接纳,而FDA审评,只是启航的第一步。
参考资料:
1.网页链接
2.网页链接
3.网页链接
3.未达主要终点仍能获批?解决不了试验设计问题,那就放宽批准条件吧……
5.其他公开资料
图片来源:药时代
版权声明/免责声明
本文为原创文章。
本文仅作信息交流之目的,不提供任何商用、医用、投资用建议。
文中图片、视频、字体、音乐等素材或为药时代购买的授权正版作品,或来自微信公共图片库,或取自公司官网/网络,部分素材根据CC0协议使用,版权归拥有者,药时代尽力注明来源。
如有任何问题,请与我们联系。
衷心感谢!
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 