


1
DNA双链断裂(DSB)修复途径
合成致死性是诱导细胞死亡的生物过程,其基于同时抑制在细胞存活所需的过程中的两种途径并行作用,仅抑制一种途径会导致细胞存活。合成致死策略已在抗癌治疗中得到广泛应用。由于一种途径可能由于转化相关变化而在癌细胞中失活,因此靶向另一种途径触发细胞死亡,同时保留健康细胞1。
癌细胞的关键特征之一是由DNA损伤的积累,包括DNA双链断裂(DSB),这是细胞中最致命的DNA损伤之一。然而,癌细胞能够通过调节其DNA修复途径存活和增殖,这可能与正常细胞中的DNA修复途径不同1。
DSB可以通过两种主要机制进行修复:BRCA1/2介导的同源重组(HR)和典型 DNA-PKcs介导的非同源末端连接(c-NHEJ)(图1)1。HR被认为是一种准确的DSB修复途径,因为它依赖于细胞周期S/G2期细胞的姐妹染色单体作为DNA合成和DSB修复的模板。因此,HR能够修复DSB,主要在增殖细胞中1。
HR修复途径由两个经典的肿瘤抑制基因BRCA1和BRCA2编码的蛋白质进行全面调节,然后募集RAD51及其旁系同源物(包括RAD51B,RAD51C,RAD51D,XRCC2和XRCC3)和RAD54以识别同源DNA模板并进行链侵袭以修复DSBs。另一方面,NHEJ是一种容易出错的DSB修复机制。NHEJ可以是分为c-NHEJ和a-NHEJ(也称为MMEJ或TMEJ)两种途径1。
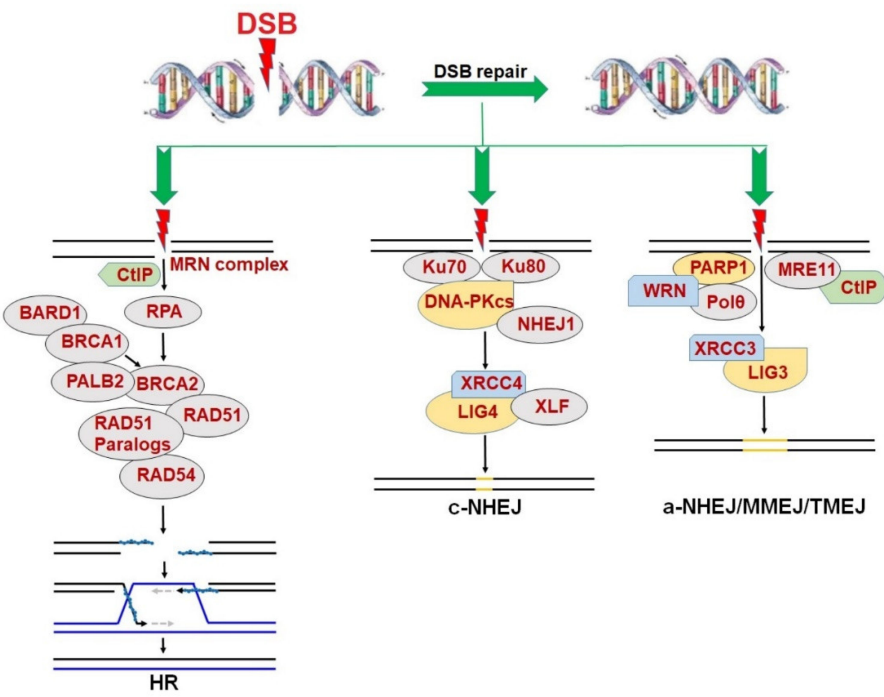
图1. DNA双链断裂(DSB)修复途径包括同源重组(HR),规范非同源末端连接(c-NHEJ)和替代非同源末端连接(a-NHEJ)/微同源介导的末端连接(MMEJ)/DNA聚合酶θ(Polθ)介导的末端连接(TMEJ)
2
PARPi在造血恶性肿瘤中的治疗潜力
最近的很多研究表明,即使在白血病中很少检测到BRCA1/2突变,PARP抑制剂(PARPi)诱导的合成致死性也可以有效地在 BRCA1/2 缺陷造血恶性细胞中起作用。
此外,诱导造血恶性肿瘤的遗传改变,例如驱动骨髓和淋巴恶性肿瘤的癌基因,包括 AML1-ETO(也称为 RUNX1-RUNX1T1)、BCR-ABL1、PML-RARa、TCF3-HLF、IDH1/2mut和IGH-MYC以及肿瘤抑制基因(例如TET2、WT1)的功能缺失突变可导致HR和/或c-NHEJ活性失调,从而使细胞容易受到PARPi引发的合成致死效应的影响(图2和图3)1。
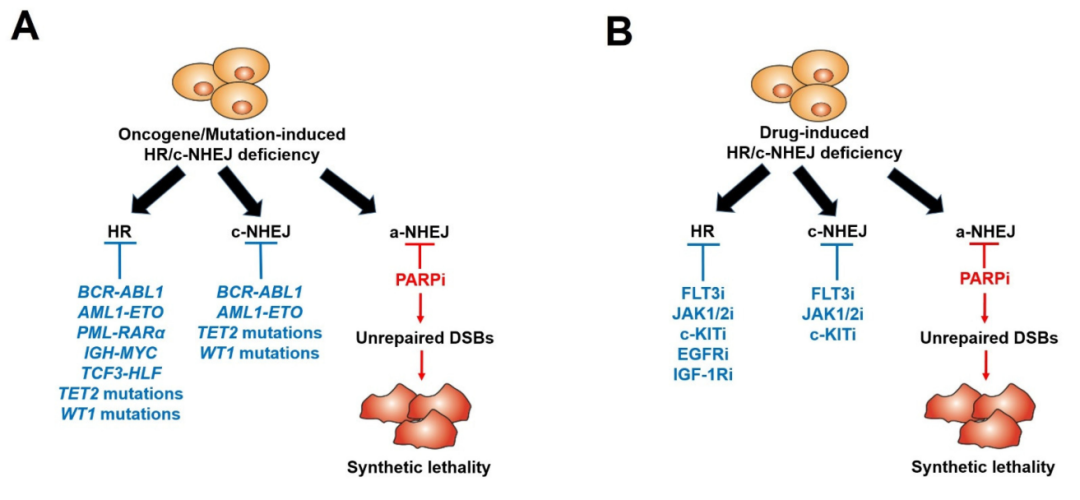
图2. PARP抑制剂在造血恶性肿瘤和其他显示癌基因/突变(A)和酪氨酸激酶抑制剂(B)诱导的HR/c-NHEJ缺乏症的肿瘤中的应用
此外,Bac等人描述了那些恶性造血细胞表达致癌酪氨酸激酶 (OTK)(例如 BCR-ABL1、FLT3(ITD)、JAK2(V617F))由于ROS产量增加自发积累高水平的氧化DNA损伤和DSB 1。
然而,OTK阳性细胞能够逃逸来自 DSB 的细胞毒性作用,这是由于增强/调节的 DSB 修复。FDA批准的特异性酪氨酸激酶抑制剂(TKi)(JAK1/2抑制剂鲁索利替尼,FLT3抑制剂奎扎替尼,ABL1抑制剂伊马替尼)对这些OTK的抑制导致急性HR / c-NHEJ缺乏症(由于BRCA1,BRCA2,RAD51和/或LIG4的下调)和对PARPi的敏感性(图2-4)。因此,TKi和PARPi的联合使用能够根除增殖和静止的恶性造血干细胞和祖细胞1。
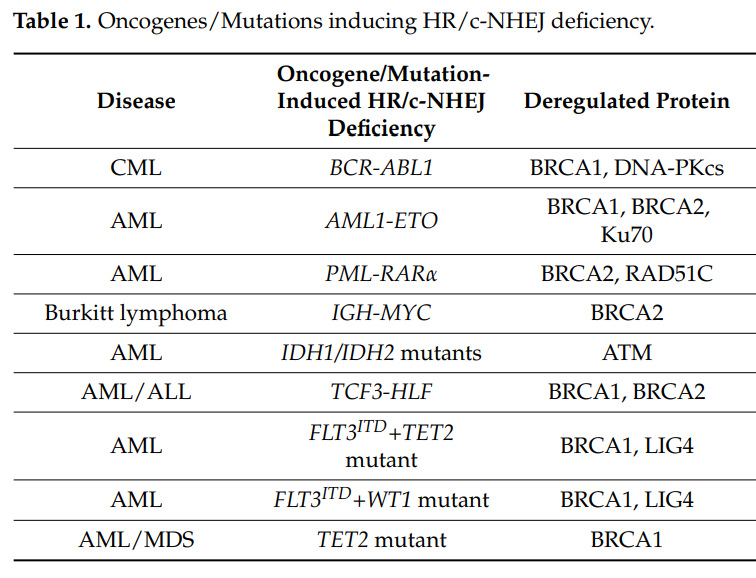 图3. 诱发HR / c-NHEJ缺乏症的癌基因/突变
图3. 诱发HR / c-NHEJ缺乏症的癌基因/突变
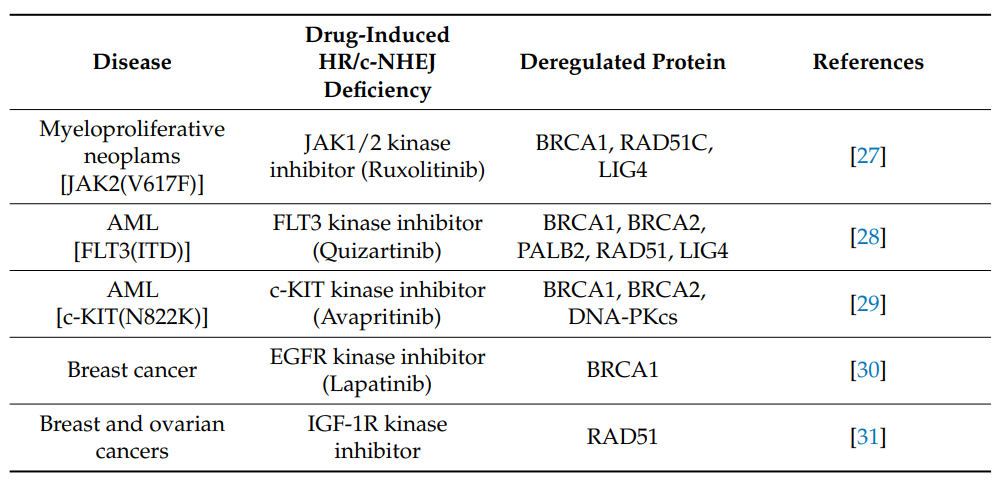
图4. 诱导HR/c-NHEJ缺乏的治疗药物
3
对PARPi的获得性耐药机制
尽管 PARPi 在 DSB 修复缺陷的癌细胞中具有很强的效力,但在BRCA1/2 缺陷和 HRD 中已经报道了对 PARPi 诱导的合成致死性的抵抗力肿瘤细胞(图5)。
PARPi的耐药性的主要原因是BRCA1/2突变癌细胞HR修复活性的恢复2。详细地说,BRCA1/2中的继发突变与原始突变导致的链终止子/移码的废除有关,导致全长 BRCA1/2 开放阅读框的恢复。因此,活性BRCA1/BRCA2蛋白表达被恢复,从而重新启动功能齐全的HR途径3,4。除了BRCA1/2中其他突变介导的耐药性外,还提出了BRCA1启动子甲基化的减少/丧失,从而恢复了BRCA1的表达而导致对PARPi的耐药性5。
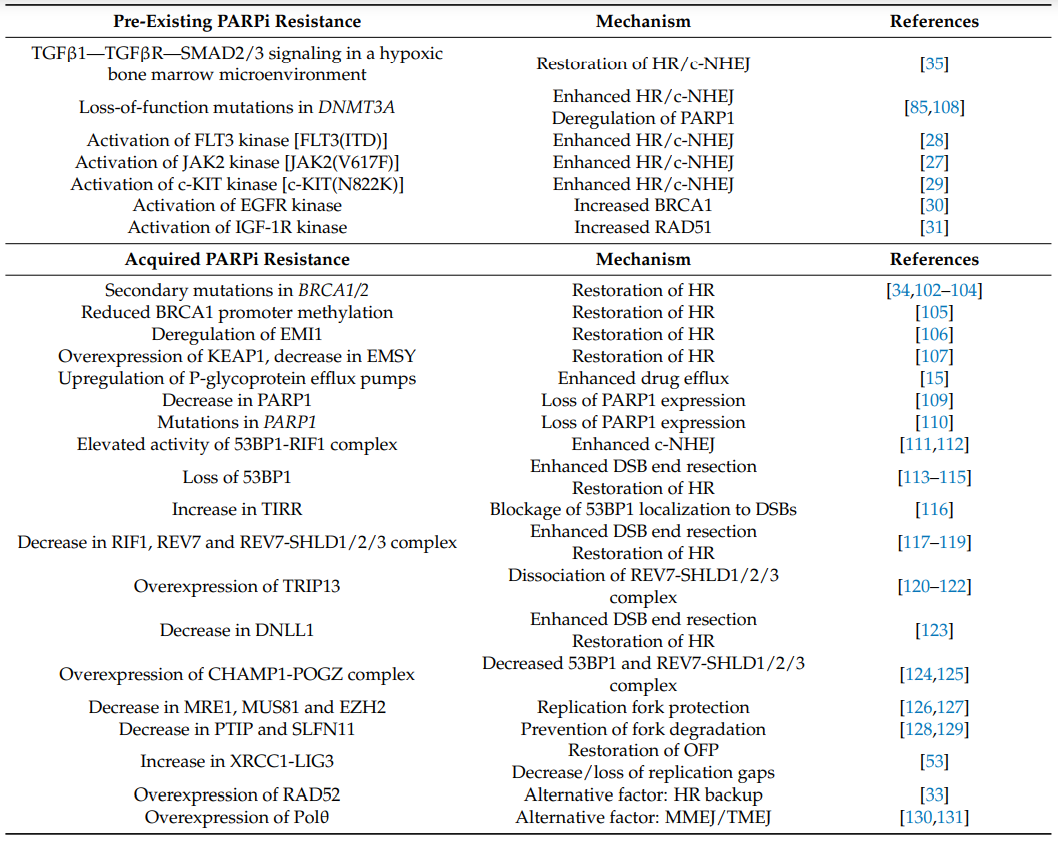
图5. 对PARPi的获得性耐药机制
BRCA1-BRCA2-RAD51轴对于同源重组修复(HRR)至关重要。EMI1调节三阴性乳腺癌(TNBC)细胞中的 PARPi 敏感性。EMI1的F盒结构域组装一个SCF泛素连接酶复合物,该复合物靶向RAD51进行降解。在一部分对PARPi难治的BRCA1缺陷三阴性乳腺癌细胞中,EMI1表达的降低会损害EMI依赖性RAD51的降解,从而恢复HR修复活性导致对PARPi的耐药性(图6)6。
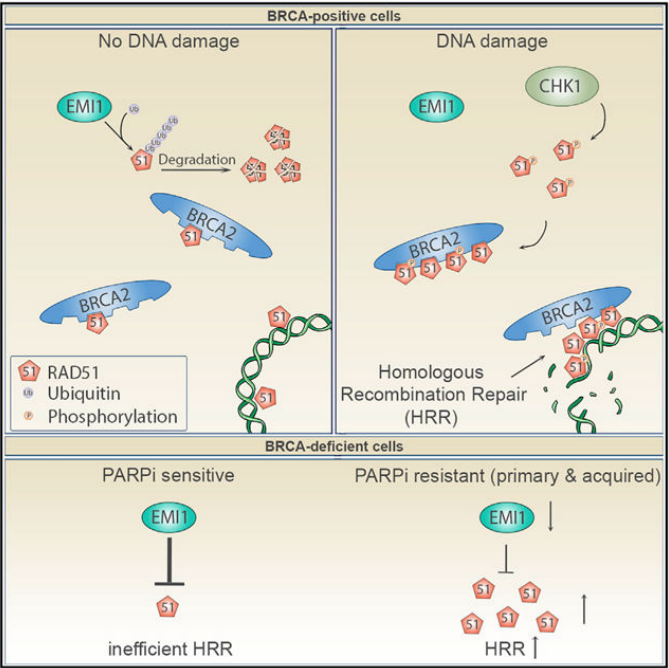
图6. KEAP1的增加导致EMI1表达的降低导致对PARPi的耐药机制
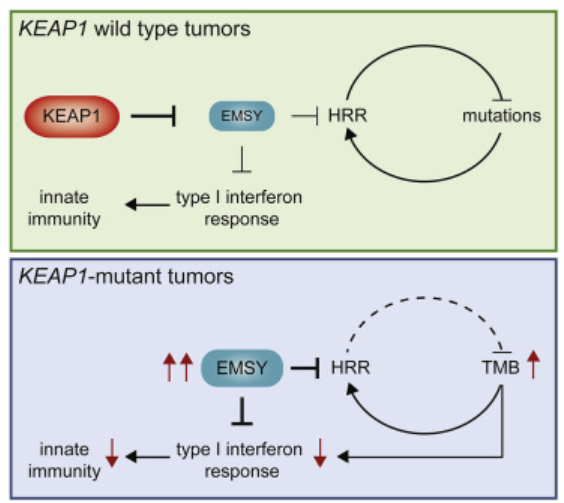
图7. EMI1表达的降低导致EMSY的减少,从而对PARPi的耐药机制
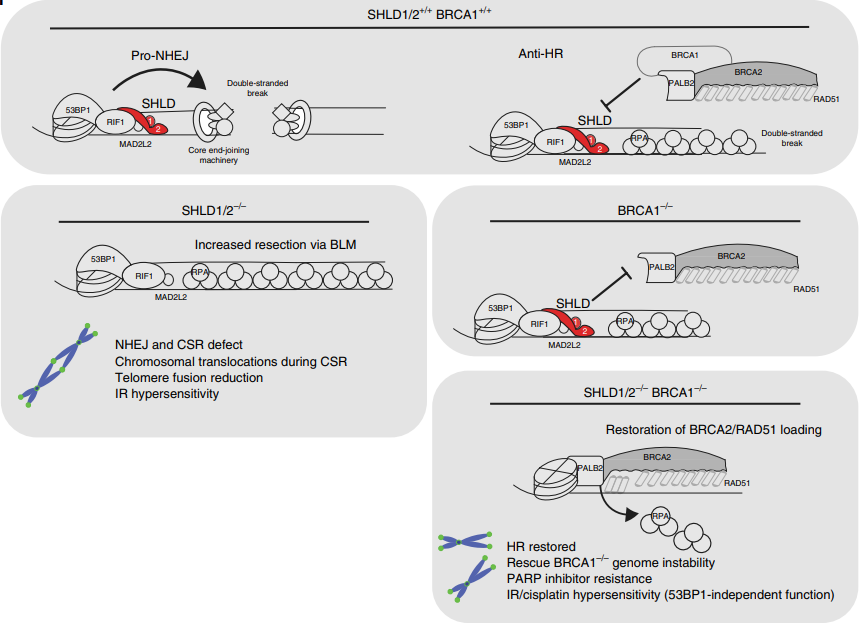
图8. SHLD1/2的敲除在BRCA1敲除细胞中导致对PARPi的耐药机制
除了在HR通路中起作用外,据报道,BRCA1和BRCA2在维持复制分叉的完整性方面发挥作用12。在BRCA1和BRCA2缺陷细胞中对PARPi获得性耐药可能是由于EZH2缺失导致MRE1和MUS81核酸酶表达降低,导致对复制叉的保护性反应(图9)12,13。
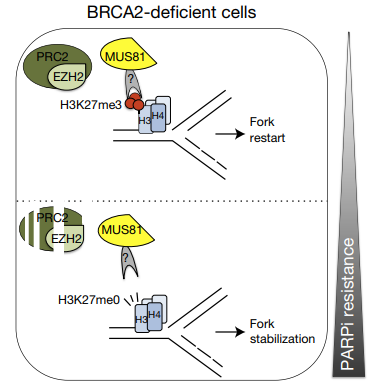
图9. MRE1、MUS81和EZH2的下调导致对PARPi的耐药机制
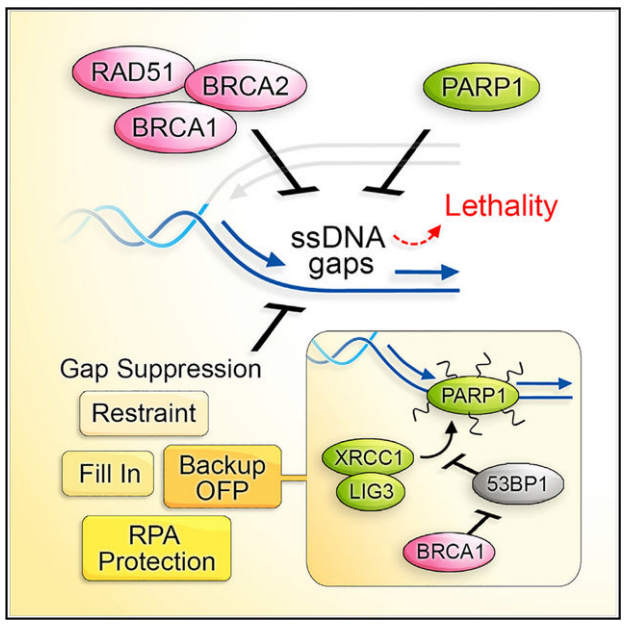
图10. XRCC1-LIG3的增加恢复OFP对PARPi产生耐药性
4
预先存在的 PARPi 耐药性机制
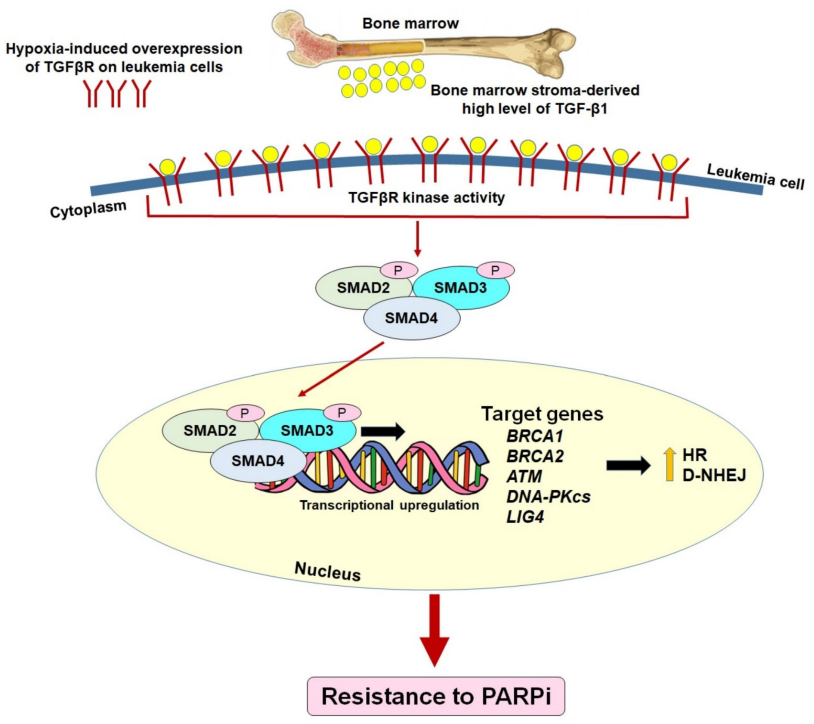
图11. 通过在缺氧骨髓微环境中激活TGF-β1—TGFβR—SMAD2/3信号传导,恢复HR/c-NHEJ缺陷白血病中的HR/c-NHEJ的PARP抑制剂耐药的预先存在机制
除此之外,还有一些其它预先存在的对PARPi的耐药机制,比如DNMT3A的突变导致功能丧失,FLT3激酶与ITD融合突变导致FLT3激酶激活,JAK2的V617K突变导致JAK2激酶的激活,c-KIT的N822K突变导致c-KIT激酶的激活,EGFR的激活和IGF-1R的激酶等都可能导致预先存在的PARPi抑制剂的耐药(图12)1。

图12. 预先存在的 PARPi 耐药性机制
5
小结
参考文献:
1.Bac Viet Le, Paulina Podszywałow-Bartnicka, Katarzyna Piwocka, and Tomasz Skorski, Pre-Existing and Acquired Resistance to PARP Inhibitor-Induced Synthetic Lethality, Cancers 2022, 14, 5795.
2.Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005.
3.Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120.
4.Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115.
5.Jacot,W.; Thezenas, S.; Senal, R.; Viglianti, C.; Laberenne, A.C.; Lopez-Crapez, E.; Bibeau, F.; Bleuse, J.P.; Romieu, G.; Lamy, P.J. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer 2013, 13, 523.
6.Marzio, A.; Puccini, J.; Kwon, Y.; Maverakis, N.K.; Arbini, A.; Sung, P.; Bar-Sagi, D.; Pagano, M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol. Cell 2019, 73, 224–237.e226.
7.Marzio, A.; Kurz, E.; Sahni, J.M.; Di Feo, G.; Puccini, J.; Jiang, S.; Hirsch, C.A.; Arbini, A.A.; Wu, W.L.; Pass, H.I.; et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell 2022, 185, 169–183.e119.
8.Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Pol_-dependent fill-in. Nature 2018, 560, 112–116.
9.Drané, P.; Brault, M.-E.; Cui, G.; Meghani, K.; Chaubey, S.; Detappe, A.; Parnandi, N.; He, Y.; Zheng, X.-F.; Botuyan, M.V.; et al. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature 2017, 543, 211–216.
10.Dev, H.; Chiang, T.-W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965.
11.Clairmont, C.S.; Sarangi, P.; Ponnienselvan, K.; Galli, L.D.; Csete, I.; Moreau, L.; Adelmant, G.; Chowdhury, D.; Marto, J.A.; D’Andrea, A.D. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat. Cell Biol. 2020, 22, 87–96.
12.Lomonosov, M.; Anand, S.; Sangrithi, M.; Davies, R.; Venkitaraman, A.R. Stabilization of stalled DNA replication forks by the BRCA2 breast cancer susceptibility protein. Genes Dev. 2003, 17, 3017–3022.
13.Rondinelli, B.; Gogola, E.; Yucel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi,R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378.
14.Cong, K.; Peng, M.; Kousholt, A.N.; Lee,W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo,J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e3127
15.Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499.
16.Wick, A.; Desjardins, A.; Suarez, C.; Forsyth, P.; Gueorguieva, I.; Burkholder, T.; Cleverly, A.L.; Estrem, S.T.; Wang, S.; Lahn, M.M.; et al. Phase 1b/2a study of galunisertib, a small molecule inhibitor of transforming growth factor-beta receptor I, in combination with standard temozolomide-based radiochemotherapy in patients with newly diagnosed malignant glioma. Investig. New Drugs 2020, 38, 1570–1579.
17.Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Pol_ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636.
18.Zatreanu, D.; Robinson, H.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.; Langdon, S.; et al. Abstract 5697: Targeting PARP inhibitor resistance with Pol_ inhibitors. Cancer Res. 2022, 82, 5697.

本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 