


正文共: 2981字 8图

我们先来看一下,D药递交申请基于的试验是怎么样的。
尽管2022年11月礼来披露了D药与A药头对头三期临床试验的结果,但上市申请早就递交,基于的是一项名为TRAILBLAZER-ALZ的二期临床试验。试验数据在2021年5月6日发表在了NEJM杂志上。
当初多入几个不就行了。
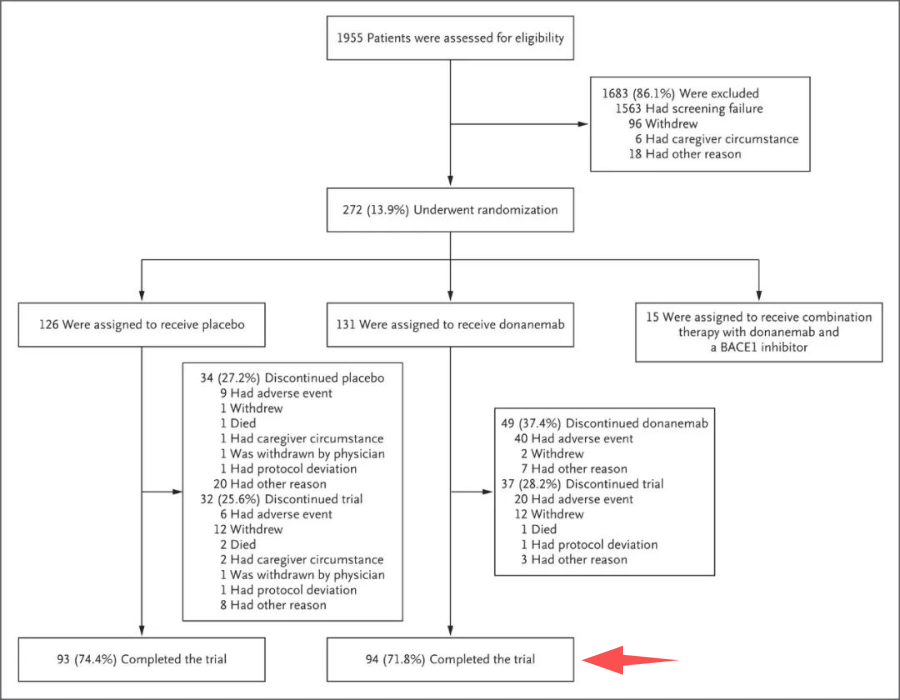
对比一下A药和Lecanemab(以下简称:L药)递交申请时给的样本量。
A药是跳过二期直接做三期,获批基于的EMERGE研究样本量超过1500例,安慰剂组、低剂量治疗组和高剂量治疗组都超过了500名患者。
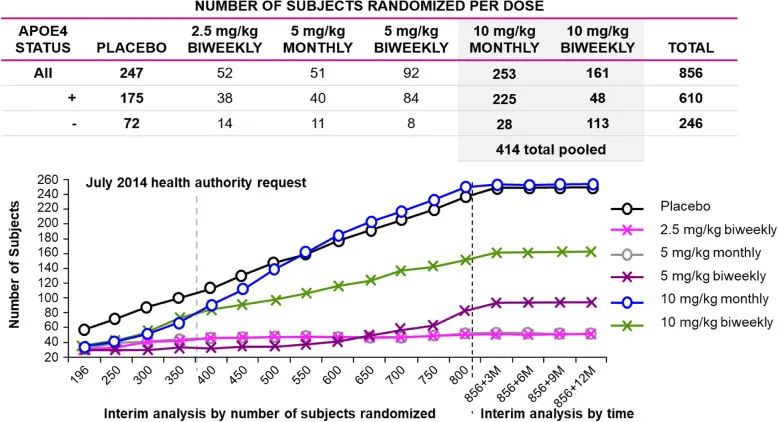
L药获批基于的二期临床数据也有超过800名患者入组,其中有610名在治疗组里。
如果要最大化药品上市成功率,与监管机构的密切沟通必不可少。
可以肯定的是,在TRAILBLAZER-ALZ试验开始前,试验方案肯定是与FDA沟通过并且获得了认可的,如果在试验开始前的沟通中,FDA已经明确表示过治疗组患者最终样本量必须大于100,那礼来肯定是全锅。
如果没有在开始前约定好,而是在审批过程中才觉得94个患者数量太少,那FDA多少得背点锅。
毕竟,在A药两项三期临床试验结果矛盾的情况下,都能召开专家咨询委员会慎重讨论,并且在专家反对的情况下仍然坚持获批,说明了FDA在阿尔茨海默病领域的开放态度。
那么,是否应当保持对同一类药品的评价统一性?是否可以在礼来设计干预方案时就表达出对于样本量的担忧?如果是在礼来方面已经递交申请后才觉得样本量太少,是否也可以召开一个专家咨询委员会讨论一下过少的样本量能否支持药品获批?
L药在出现了3例患者死亡的情况下,仍然能获批上市,那因为样本量少了6个而拒绝D药上市,多少有点“双标”。
L药的加速获批是阿尔茨海默病领域的里程碑事件,有一种观点认为L药名为“加速获批”,实为“常规批准”,因为在FDA加速批准前,其确证性三期临床试验结果已经出炉,并取得积极结果。
新药的审批是一个动态过程,监管机构对药品的态度完全可能依照现有疗法的改变而发生变化。
最典型的是突破性疗法的认定,FDA在过去两年取消了17个突破性疗法认定,有一部分原因是因为现有治疗局面发生巨大变化。
比较知名的案例是默沙东的抗丙肝“口服鸡尾酒疗法”艾尔巴韦格拉瑞韦片(商品名:择比达),曾在2013年10月的时候被FDA授予突破性疗法认定,但2014年吉利德公司Harvoni和艾伯维公司的Viekira Pak上市后,FDA就以“丙肝领域治疗迅速发展”为由,撤销了其突破性疗法认定。
不过这并没有影响其获批上市,毕竟试验做的还是一板一眼的,数据很好。
而如果L药的确证性试验已经证实了Aβ假说的可行性,FDA就很有可能对后续的抗Aβ药物都提出类似的试验终点要求,至少,是更高的试验结果要求,以“纠正”自己之前过于宽松的监管态度。
那么,礼来就是第一个“挨刀”的。
换句话说,L药打破常规地完成了“改善症状”的硬终点,有可能成为压死D药的最后一根稻草?
D药被拒跟很多药企其实没多大关系,但弄清楚责任出在哪里,避免刀砍刀自己头上,确实挺有必要。
无论是试验设计时对样本量冗余的考量、干预措施的设计,还是跟监管机构的密切沟通,负责人都要考虑全面,责任重大。
另外,研发创新药时考虑到药品开发过程中治疗格局的变化确实也挺重要,不仅关系获批与否,也关系上市后的销售额。
虎年对礼来确实是流年不利,年头、年尾各迎来当头一棒。期待兔年能有好的进展,也祝愿Donanemab能早日完成后续试验,尽早获批上市,解决阿尔茨海默病领域的燃眉之急。
1、Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, Apostolova LG, Salloway SP, Skovronsky DM. Donanemab in Early Alzheimer’s Disease. N Engl J Med. 2021 May 6;384(18):1691-1704. doi: 10.1056/NEJMoa2100708. Epub 2021 Mar 13. PMID: 33720637.
2、Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL. A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther. 2021 Apr 17;13(1):80. doi: 10.1186/s13195-021-00813-8. Erratum in: Alzheimers Res Ther. 2022 May 21;14(1):70. PMID: 33865446; PMCID: PMC8053280.
3、礼来官网
4、近2年,FDA撤销了17项「突破性疗法认定」。这张票,似乎没有想象中“值钱”
5、其他公开资料

本篇文章来源于微信公众号:药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 