
 药时代创新药BD高阶研讨会火热报名中!
药时代创新药BD高阶研讨会火热报名中!

本文是呈益投研对TPD领域历史沿革的梳理,作为呈益投资决策的参考之一。本文共3万多字,将分上下两部分在药时代宣传矩阵上独家发表。第一部分主要是UPS系统和相关药物的发现历程;第二部分涉及分子胶类TPD药物的历史以及其他蛋白降解系统。参考文献会在第二篇文末统一列出。
过去很长一段时间,科学界一直认为细胞内的蛋白是稳定不变的,即便上世纪50年代之后,科学界在蛋白代谢领域有诸多的进展,但这种共识仍旧持续到了上世纪80年代。
上世纪40年代,人体中的蛋白仍被认为是相对稳定的组分,只有极小的损耗,日常食用的蛋白只是用来提供能量,并不参与维持和补充现有体内蛋白的结构和功能。造成这种认识的原因一方面是基于科学家的经验和观察,同时科学家亦缺少相应的试验工具来对蛋白的代谢进行研究。
突破来自一位在二战期间逃离德国的科学家Rudolf Schoenheimer。Schoenheimer在美就职于哥伦比亚大学,在校期间他结识了对同位素有深入研究的科学家Harold Urey,彼时放射性同位素示踪技术也已成熟。
Urey是氢同位素氘的发现者,之后他又成功的对15N进行了富集并将之用于标记络氨酸。Schoenheimer等科学家将15N标记的络氨酸喂养小鼠,他发现只有50%的15N通过小鼠尿液回收,其余50%则留在了小鼠的组织蛋白中。这个结果表明动物体内的蛋白也在不断进行新陈代谢。但这个理论在50年代也被不断质疑,有些试验表明一些蛋白如β-半乳糖苷酶在细菌中一直保持非动态的稳定并将之扩大到哺乳动物中,此外在当时的科学环境下,Schoenheimer的蛋白动态学说不能被学界广泛接受。
直到上世纪八十年代,随着各种蛋白平衡和代谢的机制被广泛发现和完善,直至相关的蛋白降解机制的药物开始上市销售,Schoenheimer的学说才被广泛的接受,人们这才意识到他的试验是如此具有开创性。作为这一领域的早期开创者和先驱,Schoenheimer同时也是一位具有悲剧色彩的科学家,他后来患有严重的抑郁症,在1941年服用氰化物自杀。
历史的车轮滚滚向前,伴随着二战的结束,上世纪50年代,生命科学空前繁荣, 很多我们上学教材里的生命科学理论都来自那个年代的发现,最有名的当属沃森(J. D. Watson)和克里克(F. Crick)于1953年提出的DNA立体结构模型,人们对生命的认识深入到了基因层面。虽不如DNA遗传密码的发现那样伟大,但在蛋白代谢领域,新的发现也非常令人振奋,溶酶体和过氧化物酶体在那个年代被相继发现,且都和Christian de Duve 博士的“意外”研究相关。
Duve博士的研发之路可谓传奇,他是一位比利时科学家,一生获奖无数。发现和定义溶酶体和过氧物酶体两大细胞器而获得诺贝尔奖,现在广为人知的细胞活性现象“自噬”、“内吞”和“胞吐作用”都是由Duve博士命名而来。
在其研究生涯早期,Duve博士在比利时勒芬大学研究胰岛素。此时胰岛素作为降血糖药物已经由礼来制药首先研发成功并将其在1921年推入市场,但当时的胰岛素主要来自动物,在人体使用有时反而会适得其反,导致患者的血糖升高。这种现象引起了科学家的广泛关注,Duve及其团队研究并证明了礼来制药的胰岛素不纯,含有其他杂质,他们采用了胰岛素结晶纯化法使得胰岛素进一步提纯从而消除了其副作用,礼来制药基于此改进了其胰岛素的提纯和生产方式。在那个年代,Duve教授解决了胰岛素的提纯问题,且获得巨大成功。但Duve教授并没有止步于此,他想知道胰岛素提取物中是什么杂质导致了患者血糖升高?
在洛克菲勒研究所,Duve与1971年的诺贝尔奖获得者Sutherland继续致力于胰岛素杂质的研究,功夫不负有心人,他们发现了高血糖-糖原分解 (HG) 因子,这正是早期礼来胰岛素混合物导致患者血糖升高的原因,进一步证实其为胰高血糖素,他们共同对胰高血糖素的来源和作用机制继续进行了深入的研究。
此时的Duve无疑极为成功,不仅帮助礼来制药进一步纯化了胰岛提取物,并进一步证实其各种组分的作用机制。但Duve教授的目光仍不止于此,他还在继续深挖胰岛素降血糖的作用机制。在那时,一种糖代谢关键的酶“葡萄糖 6-磷酸酶”落入了他的视野中,葡萄糖 6-磷酸酶(G6p)是糖代谢(糖酵解)中的关键酶和胰岛素的下游底物,他们在研究中首先需要分离和纯化这种酶。但随后的实验他们发现,很难从细胞提取物中分离和纯化G6p,此外令Duve教授感到疑惑的是刚提取出来新鲜的G6p的活性极低,但从一些已储存五天的细胞中提取的部分的酶活性却有着很高的活性,这似乎在说明某种屏障(膜)限制了葡萄糖 6-磷酸酶快速接近底物,这种屏障亦导致了葡萄糖 6-磷酸酶较难进行分离纯化。Duve 教授进行了复杂的实验设计和验证并将这种亚细胞结构的“屏障”命名为“溶酶体”以反映它们具有水解作用的特性。随后佛蒙特大学的Alex B. Novikoff访问了 Duve 的实验室,并利用电子显微镜看到了“它”,溶酶体被发现了,单层膜包裹的酸性水解酶结构,类似细胞膜,非常美妙的结构。
但如果认为Duve会止步于此,那就错了,他随后对尿酸氧化酶发生兴趣,并紧追不舍发现了过氧物酶体,这里就不再过多赘述了。
Duve教授的一生充满了“意外”,本来想学医的他“意外”进入了胰岛素组,一系列的科学问题驱动他的好奇心,“意外”发现了溶酶体,并在之后发现并定义了过氧化物酶体。但“意外”之外,是他的兴趣和好奇心,Duve在一次采访中多次提到了意外和好奇心,这是他成功的源泉,他享受了追求好奇的带来的”意外“惊喜和成功。
Schoenheimer和Duve教授的早期研究似乎能解释蛋白质代谢和降解的部分问题,但有很多问题仍旧不能得到解答。在60-80年代,对于蛋白质代谢领域,还是处于一个争议的年代,科学界具体的问题如下:
1)一是很多实验室相继发现不同蛋白的稳定性大不相同,半衰期短则10来分钟,长的达到半个月,差了3个数量级。而溶酶体中多种水解酶会无差别的水解各种蛋白,这与不同蛋白质半衰期的巨大差异相悖。
2)另一个发现也随之而来,使用广谱的溶酶体抑制剂,对不同蛋白的降解速度的影响也不同。
3)最后一个问题,科学家们发现很多蛋白的降解需要能量的消耗,而溶酶体维持了一种酸性环境,在其中降解的蛋白质是释放能量的过程。
以上发现使得科学界越来越倾向蛋白降解还存在其他的机制,虽然在此期间科学家对溶酶体的研究也逐渐深入,对以上争议进行了勉强的解释,如蛋白进入溶酶体的机制、需要不同的受体导致其降解速率不同等等。但学界正在慢慢达成共识,一种新的维持体内蛋白代谢和平衡的机制即将揭晓,泛素-蛋白酶体系统(UPS)在80年代被科学家揭开了神秘的面纱。
ALEXANDER VARSHAVSKY(瓦尔沙夫斯基)博士是一位前苏联科学家,他在1977年通过朋友帮助来到美国,第一站就来到麻省理工MIT。在前苏联,VARSHAVSKY博士主要研究染色体结构及其对基因表达的调控。来到美国后,在麻省理工,他仍旧持续了这方面的研究。
1977年底,也就是VARSHAVSKY来到美国的当年,他读到一篇很有意思的Paper,文中介绍了一种结构比较奇特的组蛋白H2A,和普通蛋白质的氨基酸序列不同,这个蛋白拥有两个N端,一个C端。类似一个叉子的结构。很快这个Y型蛋白的短臂就被Hunt和Dayhoff鉴别出来,一种广泛存在的蛋白,早在1975年就被Goldstein发现并命名为泛素。但泛素的功能一直不为人知。这个重要的Y型蛋白如下图所示:

机会总是留给有准备的人,VARSHAVSKY博士对首个报道的泛素-H2A的结合体非常感兴趣。前面说到VARSHAVSKY博士在前苏联一直从事染色体结构的研究,他在苏联就开发了一种高分辨率的核小体分析方法,用之来分析H2A再合适不过。在MIT,VARSHAVSKY博士和他的学生开始在核小体和染色质中分离出了泛素-H2A,进一步的研究发现,泛素-H2A在转录活跃的区域较多,而在转录不活跃的区域则很少,比如着丝粒附近的异染色质。泛素似乎通过结合组蛋白调节了基因的转录和翻译,答案就要呼之欲出了。
VARSHAVSKY博士的研究主要集中在1980-1982年间,而在几乎同一时期,在以色列理工学院,Hershko和他的学生Ciechanover也发现了一种蛋白APF-1(ATP-dependent proteolytic factor 1),Hershko教授发现蛋白质在降解前会和APF-1共价结合,他们认为蛋白连接APF是他们要被降解的重要信号。
Hershko博士在70年代初曾在UCSF的Tomkins实验室当博后,当时的研究方向就是蛋白质的降解,正如我们在前文中所述,上世纪70年代,科学家们对蛋白降解领域有诸多争议,虽然溶酶体早已被发现,但溶酶体并不能解释所有的蛋白降解及代谢现象,很多实验室都发现在蛋白代谢领域有着许多用溶酶体理论不能解释的现象。
当时对于蛋白降解的具体作用机制和调控方式,科学界知之甚少,偶尔一些零星的规律被偶然发现,但可能却更加增加了学界的困扰。Hershko和Tomkins当时做研究的细胞是HTC(一种肝肿瘤细胞)中的络氨酸氨基转移酶的降解,他们发现培养基、蛋白合成以及PH都会影响这种蛋白的降解,说明了蛋白降解的可控性。最重要的是他们发现不同于溶酶体降解途径,这种降解依赖ATP,虽然类似这种研究在一定程度上说明蛋白降解存在除了溶酶体以外的体系,但总体上,在这个领域,科学家仍旧在困惑中前行。
突破来自于一种相对特殊的细胞,网织红细胞,Rabinovitz和Fisher早在60年代就对兔的网织红细胞进行研究, 网织红细胞不含有溶酶体,却可降解异常的珠蛋白,是一种绝佳的研究非溶酶体系蛋白降解系统的体系。1977年哈佛医学院的JOSEPH D. ETLINGER和ALFRED L. GOLDBERG建立了胞外的网织红细胞降解体系(cell free system)并构建使其功能和细胞内保持了一致。
Hershko敏锐了发现了这一系统的优势,cell free 系统非常有利于UPS系统中各种关键酶的纯化和特性研究。1980年Hershko和他的团队纯化并得到了APF-1,并很快将这种蛋白鉴定为泛素。
Hershko和VARSHAVSKY教授两个团队在上世纪80年代的研究可谓争先恐后,两位科学家的研究进展非常的接近。在组蛋白调控中发现泛素后,VARSHAVSKY教授之后的工作更为顺理成章,他非常敏锐的意识到需要一个细胞体系来验证之前的猜想,很“幸运”,Varshavsky教授找到了一株UPS系统异常的细胞系ts85,在那个年代没有如此众多基因编辑工具的前提下,没有crispr-cas9, 大自然创造的这些天然突变株对科研往往起到了关键的作用。后来证实发生突变的是ts85细胞中的E1,这是一种激活泛素的酶,对温度及其敏感,这是科学界第一次在细胞内观察到了泛素-蛋白酶体系统的运作。
ts85细胞内的UPS系统对温度敏感导致H2A异常,从而使细胞周期停留在G2期,目前很多UPS系统调节蛋白降解的靶点都是细胞周期蛋白和转录因子,这也许正是造物主的巧妙安排,通过分布于人体不同部位、不同类型细胞的E3来达到特异性调节转录因子的作用,
VARSHAVSKY的团队后来首次克隆了泛素的基因,并发现了触发UPS的机制“degron”。而Hershko博士也先后发现了泛素系统的三种重要的酶,我们现在熟知的E1、E2和E3连接酶。
VARSHAVSKY和Hershko博士分别在各自的领域观察到了泛素蛋白系统,很有意思的是,两位博士的研究领域和背景看起来不太相干,一位多年深耕蛋白降解领域,另一位是通过多年研究染色质功能发现泛素对其的调节。科学在这一时刻交汇,非常的美妙,让人回味无穷。这也说明了这一系统复杂精确并广泛的存在于体内,之后的发现也证明了这一点,泛素对蛋白的降解在哺乳动物体内几乎无处不在,无时不在。
Hershko和VARSHAVSKY博士对UPS系统的发现和作用机制都由深入的研究,他们从各自的领域,用各自擅长的工具对UPS做了深入的阐述,相同是的,发现都来自他们对自己领域的多年研究和积累,他们都遇到了对各自突破的重要细胞网织红细胞和ts85,一位在蛋白酶催化层面阐述UPS的机制,一位在染色体层面深究UPS对转录翻译功能的影响。并且他们的工作都发生在70年代末80年代初,彼时基因重组技术和产品也层出不穷。2004年Hershko及其团队另外两位成员获得诺贝尔奖,遗憾的是VARSHAVSKY博士与诺贝尔奖无缘,令人遗憾,但从另一个角度讲, VARSHAVSKY博士多年在蛋白降解领域,而Hershko的发现虽然和H2A密切相关,但他的工作可能更加的“意外”。无论如何在这一系统的研究上,不管是先驱Schoenheimer、还是VARSHAVSKY和Hershko,多年的积累、日复一日的追踪和敏锐的判断,甚至一点运气,都为他们的成功奠定了坚实的基础。
UPS系统最后一个重要的工作就是蛋白酶体机制的阐明,蛋白酶体各种亚结构和功能在1988年被美国科学院院士Alfred Goldberg命名和阐述。
80年代后期直到90年底,UPS系统的研究日益成熟,科学家们不断的完善了系统中的细枝末节,在两位重量级教授的框架下不断的前行,并持续完善人体蛋白质代谢的拼图,这些工作目前仍在进行中。
在90年代到新世纪,基于这个系统的药物研发也开始萌芽并开花结果,我们下面把目光从基础研究稍微前移,UPS应用于药物开发并造福患者的时代逐渐拉开了帷幕。
现在我们都知道在UPS系统中,8种E1活化酶,40种E2,以及上百种E3调控着人体的各种蛋白代谢,这些复杂的酶互相配合,在人体不同的部位和不同的活性,保证了靶向降解蛋白的特异性。涉及细胞周期、凋亡、细胞活性等众多细胞的功能获得和丧失。科学家们在以上领域也开始不断发现UPS新的功能和作用机制。
在科学界对UPS的系统研究中,蛋白酶体抑制剂发挥了重要的作用。一方面是抑制蛋白酶体对整个泛素、各种级联酶以及目标降解蛋白的机制研究至关重要。另一方面,通过蛋白酶体抑制剂,人们对蛋白酶体的功能和结构也逐步有了深入的认识。
最初科学家们需要合成蛋白酶体抑制剂并将之作用科学研究工具,这些抑制剂一般含有2-3个氨基酸短肽和羧酸作为“warhead”, 短肽是用来模拟蛋白酶体结合的底物蛋白,羧酸一般是硼酸类,在蛋白酶体生理PH情况下带负电荷,以上特性主要用来模拟水解酶的结合底物从而对蛋白酶体产生抑制作用。早期作为基础科研研究工具的蛋白酶体并没有考虑成药性,如针对蛋白酶体20s亚基的特异性、抑制效力以及体内的代谢稳定性等。
除了一些天然发现的蛋白酶体抑制剂,科学家对蛋白酶体抑制剂的设计思路都是沿着短肽和羧酸结构的母核来进行,这也是蛋白酶抑制剂早期的通用结构和化学家们优化的基础。
早在1990年,哈佛大学的Tom Maniatis和Alfred Goldberg就对蛋白酶体抑制剂治疗艾滋病伴随的肌肉萎缩症开展了早期研究,这个研究的基础是这类病人中UPS系统异常活跃,从而导致了肌肉发生萎缩。Goldberg在1993年作为科学创始人成立了MyoGenetics公司去开发蛋白酶体抑制剂,大量的生物学的数据支持只要抑制病人体内的UPS系统会对肌肉萎缩产生治疗效果,因此成立之初,MyoGenetics公司准备研发蛋白酶体抑制治疗艾滋病引起的肌肉萎缩。在理论可行的基础上,MyoGenetics需要产业界的专家来支持其药物的研发,随后药物化学家Julian Adams加入开始领导公司的业务。Julian Adams之前在勃林格英格翰,主导了抗艾滋药Viramune(一种病毒转录酶抑制剂)的研发,Viramune1996年在美国获批上市。
Julian Adams的新药研发经验无疑给是吸引Goldberg邀请其加入的重要因素。果然Adams不负众望,1994年,Julian Adams就找出了一种新的蛋白酶体抑制化合物:硼酸二肽缩合物MG-341, 与Dana-Farber Cancer Institute(DFCI)合作,MyoGenetics对MG-341的活性进行了一系列的测试。细胞和动物数据显示,MG-341在多种实体瘤和血液瘤细胞中有效,但同时也发现了不少的毒副作用,后续这个新的蛋白酶体抑制剂被考虑应用于抗肿瘤。MyoGenetics公司后续改名为ProScript,同时MG-341在后续的研发代号也改名为PS-341,1999年ProScript后来被千禧制药(Millennium Pharmaceuticals)收购,经过一系类临床试验后,2003年PS-341被FDA批准用于复发难治多发性骨髓瘤的治疗,这就是我们熟知的硼替佐米。
多年后Adams在千禧制药的同事John Maraganore回忆起硼替佐米的研发经历,John Maraganore现任Alnylam公司(现在众所周知的小干扰RNA公司)CEO,他讲到:当时大家都认为抑制蛋白酶体抑制剂会无差别抑制蛋白酶体,这可能会导致病人死亡,但Adams却一直坚信其巨大的抗肿瘤潜力。Adams成功了,蛋白酶体在虽然在生物学机制上的广泛生理作用让生物学家对其抑制剂有一定的疑虑,但在抗肿瘤领域,药学专家在平衡效果和副作用后的决定让硼替佐米成为了千年制药最有价值的资产,2008年,武田以88亿美金收购了千年制药。
1992年,也就是Goldberg在哈佛着手准备开发蛋白酶抑制剂的同时,Craig Crews刚在哈佛获得了博士学位,他是化学背景,后来获得生物化学的博士学位,之后一直在耶鲁大学的化学和药理系任职。
直到90年代后期Crews教授在耶鲁仍在寻找好的科研项目,那时的药学家通常会在植物、土壤或海洋中寻找天然的疗效分子,一天他在浏览抗生素杂志时,一个分子引起了他的注意。这是一个天然的细菌产物:环氧霉素(epoxomicin),一个四肽结构在其末端有个不同寻常的环氧酮。这个化合物是百时美施贵宝(BMS)在东京的研发团队发现的,后续测试后发现其在体内有一定的抗肿瘤活性,但BMS的东京团队当时并没有搞清楚这个化合物的作用机理,为什么它有抗肿瘤活性。后续的研究也由于BMS东京的研究院的关闭而搁浅,这导致要获得环氧霉素的样本化合物都存在一定的问题。
Crews教授对环氧霉素非常感兴趣,作为化学家他敏锐的觉察到epoxomicin的环氧酮具有被亲核试剂攻击的特性,Crews认为epoxomicin的环氧酮很可能靶向了某种蛋白,这很可能是其抗肿瘤的基础。为了获得足够数量的epoxomicin用于研究,Crews和他的学生开发了合成epoxomicin的路线,在获得足够量的epoxomicin后,他们利用生物素标记法,计划钓出epoxomicin结合的蛋白。结果很快出来了,他们钓出的蛋白就是蛋白酶体。当时Crews教授和Adams在开发硼替佐米面对的问题一致,当时很多生物学家认为抑制蛋白酶体会导致对正常细胞的伤害,因为蛋白酶体对维持细胞的活性至关重要。但Crews教授认为肿瘤的蛋白代谢相比正常细胞更为活跃,在针对肿瘤细胞的蛋白酶体抑制剂存在一定的剂量窗口,可以一定程度上抑制肿瘤细胞,而对正常细胞副作用可控。而化学家出身的Crews也敏锐的意识到epoxomicin还有很大的改造潜力,Crews决定继续推进epoxomicin项目,并计划将之推向临床。
Crews开始和德国Max Planck生化研究所的Robert huber展开合作,Robert huber是一位鼎鼎大名的结构生物学家,他因研究细菌光合反应中心的三维结构获得1988年诺贝尔奖,Crews和huber合作通过解析epoxomicin与蛋白酶体结合晶体结构阐明了epoxomicin特异性结合蛋白酶体的机理。他们发现与epoxomicin结合的蛋白酶体的苏氨酸残基,正好处于蛋白酶体亚基的N-末端,而其他蛋白酶则不是,epoxomicin这种特性,导致它只和蛋白酶体结合,而不与其他蛋白酶结合,从而有着更好的选择性和应用潜力。
解决了Epoxiomincin的作用机制后,Crews教授开始对Epoxiomincin的结构进行改造,对于化学生物专业出身的Crews来说,这简直是轻车熟路,在对Epoxiomincin改造后,Epoxiomincin衍生物的抗肿瘤效果进一步增强,在众多的衍生物中,他们得到了YU-101, YU-101的对蛋白酶体的抑制效果不仅要好于天然的Epoxiomincin,甚至要好于千禧制药的bortezomib,后者正是我们之前提到的MG-341。
在上个世纪90年代末,Crews教授手握YU-101,踌躇满志之时,但现实的冷酷让他和YU-101放慢了脚步。
新药研发在当时被称为穿越“死亡之谷”,在上世纪90年代到新世纪来临之初,biotech的数量远不如现在,能够投资biotech的VC机构就更少了,现在知名的投资机构如Flagship1999年才成立,比较活跃的投资机构也仅限Atlas等老牌的欧洲VC。在美国,虽然1999年VC在生物医药的投资翻了一番达到了40多亿美金,但2000年之后直到2002年VC在Biotec领域的投资额一直都在衰退,直到2004年VC在biotec的投资才又达到30多亿美金,远低于现在上百亿的投资规模。而在学界,很多学者也都认为新药研发难度很高,例如Scripps海洋所的教授William Fenical,他曾经创立过两家公司,经验非常丰富,但William教授在当时也认为,如果大的制药公司没有兴趣,化合物再好也永远不能获批上市。他认为创新药的风险非常之高,是一种非常残忍的商业模式。而斯坦福的Paul A. Wender教授,即便他有过三家biotech的创业经历,他仍然对创业保持了敬畏,他认为做创新药,需要很多的业内资源,很多不同经验、技能和背景的人帮助去穿越创新的“死亡之谷”。这个历程中,需要好的化学家、药理学家、生物学家和临床专家的默契配合,需要考虑投资、专利,远远超出一个人的能力范畴。
Paul A. Wender教授的话至今仍给我们很多启示,虽然他的看法是在biotec比较低迷的背景之下,当时生物医药的创业环境和活跃程度都远不如现在,但仍值得我们认真思考,尤其是当下国内创新药整体大环境遇冷之时。
言归正传,即便是要穿越“死亡之谷”,Crews教授必须迈开第一步,他花了数个月在投资公司穿梭,但却一直不能锁定投资,直到2003年,市场回暖之际,Crews教授的努力终于得到回报。转折的发生也得益于2003年3月,全球第一个蛋白酶体抑制剂bortezomib(Velcade)获批上市了。但bortezomib存在一定副作用,主要是患者的周围神经疼痛。Crews敏锐的意识到bortezomib的副作用来自其硼酸,他认为YU-101的环氧酮可以消除这个副作用,他告诉投资公司他拥有比bortezomib更好的蛋白酶体抑制剂,这次他成功了。
之后Crew和Raymond J. Deshaies一起成立了Proteolix公司来开发新一代的蛋白酶体抑制剂,为了进一步改造YU-101, 使其从实验室分子变成真正的药物,他们合成了100多种类似物进行筛选,最终在YU-101的基础上引入吗啉环,增加其溶解性,这就是carfilzomib,全球第二个蛋白酶体抑制剂卡非佐米。2009年 Proteolix公司被Onyx收购,其后期临床主要由Onyx推进,卡非佐米在2012年获批上市。
尽管carfilzomib后期主要由Onyx的临床药学团队推动,但Crews给实验室创业带来了希望,之前对于实验室教授创业的偏见和争论也没有停止,产业背景的Adams认为Crews的工作非常完美,但他同时也认为,独立的发展和独立资金供给才能说明一个产品成功穿越了“死亡之谷”,后期Onyx对carfilzomib的成功也至关重要。但不可否认,Crews教授是少有的对制药行业有深刻认识的科学家,他不是那种专注于基础研究的学者,在很早就有和大型公司合作的经验,不论是其早期与BMS引入并改造的Epoxiomincin,还是后期与新基、葛兰素史克(GSK)等大型药企在PROTAC领域的合作,他是少有的全方位跨界学者。
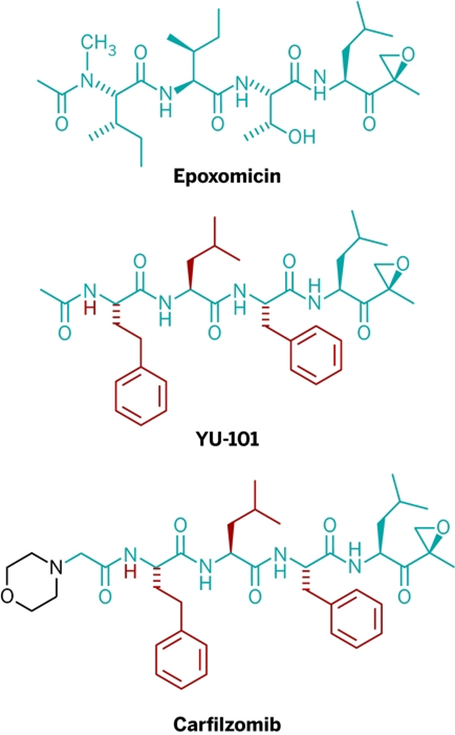
卡非佐米的改造历程
在一起创立proteolix公司前,Crew和Deshaies教授的合作事实上早就开始了,他们的传奇远不止于此。现在大家都熟悉的PROTAC在Crews和Deshaies的推动下登上舞台并逐渐引起广泛关注,但其实早在2001年,他们就已经提出了PROTAC的概念(proteolysis-targeting chimaeras)。

图片来源:298 | NATURE | VOL 567 | 21 MARCH 2019
以上来自Crews实验室官方的一张原理图,crews教授在各种场合都用到这张图,包括他实验室的官网以及他对外讲座的各种PPT。
直到现在,PROTAC都是创新药领域最热门的领域,代表并正在引领了下一代小分子药物的方向,无数的科研人员和资本都在不断地投入到这个领域中。
已知最早的关于类似PROTAC分子的公开描述是位于美国马里兰的一家生物技术公司 Proteinix, Proteinix的两名科学家John Kenten和Steven Roberts于一个1999 年提交的专利中提出同时连接目标蛋白和连接酶,从而利用蛋白降解系统消除不需要的蛋白。但Proteinix公司内部对此并没有太大的热情,认为这种方法使得药物复杂化,所以Proteinix一直没有采用这个方案。同时在美国边境的度假胜地Semiahmoo bay,另一对科学家也在考虑同样的想法,1998年,在华盛顿Semiahmoo bay的一次研讨会上,Deshaies在Crews的海报前停下来听他谈论使用小分子将两种蛋白质连接在一起。Deshaies是加州理工学院的一名生物化学家,对泛素连接酶的有深入研究。人类基因组编码了大约600个泛素连接酶,它们需要与其他蛋白质形成复合物才能发挥功能。大约一年前,Deshaies团队发现了一类含有250种泛素连接酶的蛋白家族,他和Crews有太多共同的想法和理念,整个周末,Crews和Deshaies都在谈论这个话题,在他们分开时,两人已经达成共识,他们计划一起去寻求资金帮助来实现这个想法。
而在当时,正如我们前面介绍,Crews正在开发一种与PROTAC作用相反的药物蛋白酶体抑制剂,通过阻断了细胞中的泛素系统,导致蛋白质积累到危险水平并最终引发细胞死亡。Crews认为UPS系统相反的一面,即利用蛋白酶体降解蛋白和抑制蛋白酶体一样,也会有所作为,后来的事实也证实了Crews的论断。
Crews和Deshaies都来自高校研究机构,在首次设计PROTAC并验证其概念时,他们也寻求了产业界的帮助,这次他们找到了新基,后者在2001年也开展了蛋白平衡技术平台的研究。2001年,来自耶鲁(Crews博士)、加州理工(Deshaies博士)以及产业界的新基公司(Mercurio博士)等一起发表了一项研究,他们使用蛋白降解嵌合体(PROTAC)成功的降解了一种名为 METAP2的蛋白,PROTAC头一次进入了人们的视野。

Protac-1设计主要基于Deshaies教授过往的研究成果,如下图红框中所示SCF
我们可以把Crews等人合成的第一个PROTAC分子叫PROTAC No.1, (Protac-1 ),但Protac-1这个结构很难说是一种药,甚至不能被成为一个分子。它是一个复杂、臃肿的多肽和有机化合物的结合体,很难透过细胞膜,Crews教授的试验一开始只是在非洲一种蟾蜍的卵提取物中(胞外)来验证它的降解功能,Protac-1最多可以被认为是一种为了科研而设计的概念验证工具。
SCFβ-TRCP(一种E3泛素连接酶)结合的肽段来自Deshaies教授之前的研究,而其余的分子和linker则是Crews教授等设计完成。有一个很重要的点需要说明,天然的SCFβ-TRCP的降解底物是IkBα,而不是文章中的MetAP-2. 这个突破非常重要,PROTAC可以成功利用天然E3降解研究者想要降解的蛋白,另一词语可以更传神的形容这个过程:劫持(hijack),PROTAC通过劫持E3来降解目标蛋白的概念首次得到验证,万里长征开始迈出了第一步。
Crews马不停蹄,目标就是小分子E3配体,经历了多次的实验和研究,2012年,也就是卡非佐米上市当年,Crews实验室报道了一种新的小分子E3配体,主要针对VHL,这使得Crews教授有足够信心,是时候用PROTAC来做真正的药物了。
VHL是一种抑癌基因,VHL的基因异常会引起一些肿瘤驱动基因过度表达,从而会导致一些罕见的肿瘤。VHL的抑癌机制是通过降解一种转录因子HIF-1α,而HIF-1α在基因层面会引起一些生长因子的过度表达,如促学管生成生长因子、血管内皮生长因子VEGF、glucose transporter、GLUT1以及红细胞生成素等。所以VHL其实是一种调节HIF-1α的E3。
在正常情况下,HIF-1α呈现组分表达,其含量一般很低,在正常细胞中HIF-1α被脯氨酰羟化酶羟基化,从而被VHL识别后降解。但在低氧环境下,HIF-1α的羟基化不能实现,从而导致其大量积累。2002年,牛津大学的Chris Pugh教授的团队发表了一篇文章,证实含有HIF-1α氧依赖性结构域的一段多肽可以和HIF-1α竞争性的结合VHL,Chris Pugh一直致力于缺氧对机体生理影响的机制研究,他最新的paper也是关于缺氧对免疫影响的机理性文章。本世纪初,科学家已经发现HIF-1α的降解就极度依赖氧的活性,Chris Pugh教授在此基础上对HIF-1α氧化降解机理和缺氧后机制做了进一步的研究,虽然这只是Pugh教授在缺氧后生物学机制的部分成果,对于巨大的后续转化和应用价值Pugh教授并无太多关心。但Pugh教授无心,而Crews教授深受启发。他敏锐的意识到可以利用HIF-1α中被氧化的羟基脯氨酸作为起点,来设计VHL的小分子配体。工作很快就开始进行了,这次的合作对象当然也是自牛津大学。
后来的工作对Crews团队就是轻车熟路了,他们设计并合成了一系列的羟基脯氨酸类似物,然后就是VHL结合的晶体结构解析,然后化学家们继续优化设计,把当时能用的工具统统用上,对于Crews教授来说,一切都是信手拈来,水到渠成。最终筛选到一个合适的VHL配体,大致结构如下:
尽管最早发现的E3小分子配体是nutlins,但它的分子量相比羟基脯氨酸更大,接近600的分子量让化学家很难有空间去设计PROTAC,如下,在E3小分子寻找和设计的道路上,Crews教授找的VHL配体已经非常的不易。需要说明,以上寻找涉及大量化合物验证核筛选的工作,Crews教授做了非常多的工作,精确到一个分子的差距,通过高通量筛选,高通量的合成目标化合物,也得到了来自GSK科学家的帮助,PROTAC在向成药的路径上又前进了一大步。
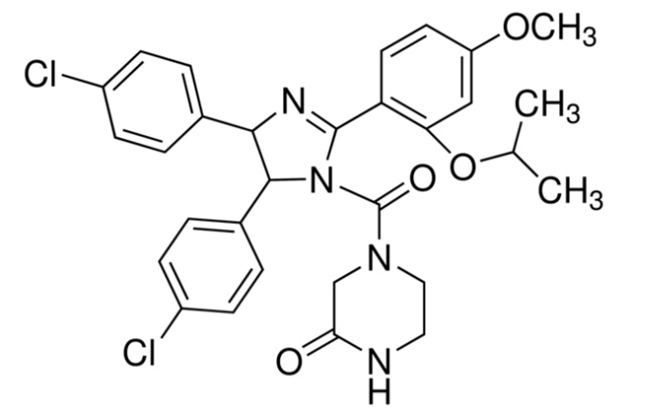
nutlins结构
但Crews教授并不是唯一专注蛋白降解药物研发的科学家,在大概同一时间,在波士顿的Dana-Farber研究院,James Bradner读到一篇文章,作者是来自东京工业大学的Hiroshi Handa, Hiroshi Handa当时刚刚开发了一种高通量筛选靶蛋白的方法,而对于臭名昭著的沙利度胺,当时科学界并不知道它的具体致畸机理,于是Hiroshi Handa采用钓鱼的方式,用沙利度胺做饵,来研究沙利度胺在细胞内的结合蛋白,最终发现沙利度胺和一种E3连接酶cereblon相结合。后续的研究进一步证实沙利度胺的正是利用E3连接酶的泛素化机理产生各种生理作用。而在当时新基的沙利度胺和来那度胺两个能与cereblon结合的化合物都已上市销售,用度胺类药物作为E3配体最为切实可行,考虑以上James Bradner认为可以利用度胺类E3配体来开发相关的小分子降解剂。
于是在2015年,Crews、Bradner以及之前在Crews实验室的英国访问学者Ciulli分别发表了三篇报道小分子PROTAC的文章,证实了PROTAC分子在细胞和动物实中同样有效。
先说Crews教授2015年的文章,由Crews教授在耶鲁的团队和GSK合作发表,需要说明的是在整个近30人的实验组成员中,GSK派出了约15人的团队,涉及三个独立的部门,涵盖了合成、分析和药物设计。GSK 2012年收购的Cellzome也涉及其中,Crews教授敏锐的嗅觉往往让他在很多院校教授的基础研究中发觉向药物应用转化的机会,同时他也非常善于寻找产业方的合作伙伴。从院校到产业的合作成就了Crews教授不断完成新的成就,开创新的领域。
Crews和GSK的科学家在2012年Crews教授筛到的VHL配体的基础上,通过linker链接了目标蛋白的配体,他们选择的蛋白是ERRα和RIPK2,分别设计了protac-errα和 protac-RIPK2,在体外细胞和体内动物实验中,靶蛋白都被特异的降解了,而且在动物体内也没有观察到明显的副作用。这是首次在细胞和动物水平报道的小分子双功能降解剂。尽管之前也有小分子的双功能降解剂bestatin,主要针对的E3是IAP, 但其配体并不是特异性的和IAP结合,从而限制了其在细胞中的正交性和独立反应,导致其在实验工具和未来成药方面有不小的局限。
2015年,Crews团队找到的小分子PROTAC的突破主要如下:
1)首次利用小分子PROTAC在体外和体内实现了有效、特异和可逆的目标蛋白降解。
2)理论上,和更早期出现的siRNA技术和2013年问世的crispr-cas9技术相比,PROTAC理论上在细胞层面可对多种蛋白的含量进行随意的调控,siRNA和crispr-cas9则分别在RNA和DNA层面进行调控。
3)使得许多之前不可能成药的蛋白实现可成药性,大大拓宽了可成药蛋白的范围。
相比传统抑制剂的占位驱动,事件驱动的PROTAC分子的效用以及成药性受到配体、linker长度以及整个组合分子特性的影响,需要化学家、结构生物学家以及药理学家更多的工作和精细的调控来实现。
除了Crews外,前文提到的在Crews实验室访问的英国学者Alessio Ciulli回国后也继续了PROTAC的相关研究,同样选择了VHL的小分子配合,靶向蛋白则选择了BRD4, 与BRD抑制剂相比,低剂量PROTAC可选择性的降解BRD4,并且实现了细胞内的降解。
James E. Bradner教授的策略则不同于Crews和Ciulli教授,前文提到过Bradner意识到邻苯二甲酰亚胺类结构的药物,如沙利度胺是E3 CBRN的结合蛋白,他意识到这是一种绝佳的E3配体,而且它的分子量更小,利用这个特性,Bradner教授也设计了一系列基于度胺类配体的PROTAC,和Ciulli教授一样,他同样选择了BRD4,但为了验证基于度胺类药物的的适应性,Bradner教授也做了胞质信号蛋白 FKBP12的双功能降解子。
三位教授的工作拉开了PROTAC研发的序幕,分子式结构如下,后续很多学者开始各种PROTAC研发的热潮。
Crews教授 PROTAC:
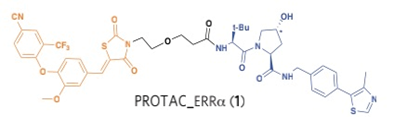
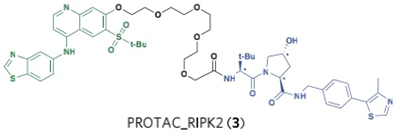
Ciulli教授 PROTAC:
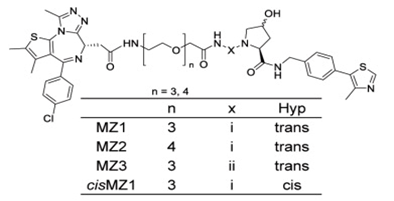
Bradner教授PROTAC:

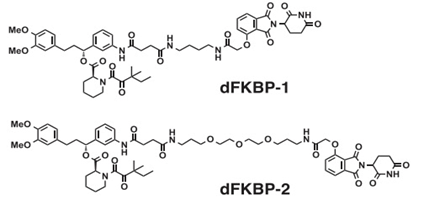
Crews教授和Bradner教授分别利用VHL和Cereblon降解了不同的蛋白,在此基础上,两位教授搭建了HaloPROTACs和dTAG的平台,研究者们使用这两个平台在实验室中用于降解目标蛋白,从而进行科学研究,类似之前的siRNA和Crispr工具。但和siRNA和CRISPR一样,真正的挑战是将科研工具转化为临床药物。PROTAC虽然比传统小分子面临的成药难度更大,但相比对基因层面进行改造的工具siRNA和CRISPR,其靶向性、成药性相对简单,拥有成本低廉,潜在可口服的优势。而siRNA和CRISPR的大规模应用则需要载体技术的突破以及基因层面可控的副作用。在蛋白层面进行调控的PROTAC和在RNA、DNA层面进行调控的siRNA和CRISPR一样,拥有极高的想象空间。
此时,Crews教授创立的ARVINAS已经成立并运营了两年,Bradner教授2016年加入了诺华,而Crew教授之前的合作伙伴,2001年和Crew教授共同提出PROTAC的Deshaies则在2017年进入了安进,负责安进这个领域的研发。
人们对PROTAC的疑虑主要来自于是否能够在人体中发挥作用,目前设计的PROTAC的分子量达到了近1000,而一般小分子药物的分子量不到500,即便如此,很多PROTAC仍可以自由的进入细胞,Crews教授认为可能细胞把PROTAC分子当做恰好挨在一起的两个小分子来识别。ARVINAS的副总裁Taylor认为:人们应该抛弃先入为主的观念,认为小分子药物的分子量小于500,目前也有大于平均水平的小分子药物。PROTAC与占位驱动的的抑制性小分子相比,他们不需要完全占领靶蛋白的活性位点,PROTAC只需要结合蛋白上一个小的角落、沟槽甚至一个缝隙就能发挥作用,因此其成药性与过往的小分子不可同日而语。学术和产业界逐渐发现了更多的证据,伦敦肿瘤研究中心发现了一种小分子,他们与一些转录因子结合的位点也并不是其活性中心;Deshaies表示安进也通过PROTAC在体外和体内有效的降解了之前不可成药的靶点;而Arvinas则宣布在体内利用PROTAC已经把老鼠脑部的tau蛋白降解了50%。
除了全新的靶点,PROTAC也可提高现有抑制剂的有效性,Ciulli教授认为,PROTAC正在改变行业规则,它们的潜力无限。2019年Avinas公司的ARV110和 ARV471启动一期临床试验。大约2年后,2021年7月23日,辉瑞宣布将以6.5亿美元首付、3.5亿美元股票认购,加上14亿美元潜在里程碑付款收购ARV-471部分权益。PROTAC正在飞速的向着最终的成药进发,极有可能改变整个小分子药物的领域和科学家之前根据抑制剂积累的小分子成药性规则。
本文为原创文章,不代表药时代的观点,版权归作者/拥有者所有。
本文非商用,不具有任何商用、医用、投资用等方面的参考价值。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频、字体、音乐等素材或为药时代购买的授权正版作品,或来自微信公共图片库,或取自公司官网/网络,
根据CC0协议使用,版权归拥有者,药时代尽力注明来源。
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!
药时代创新药BD高阶研讨会火热报名中!
本篇文章来源于微信公众号:药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权










 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 





