

重磅直播,精彩不容错过!


遵循ICH规则是国际化唯一路径
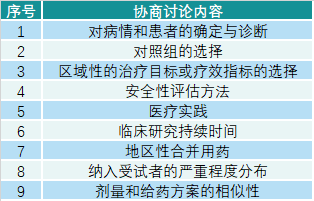
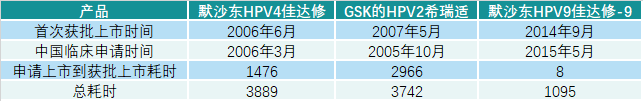
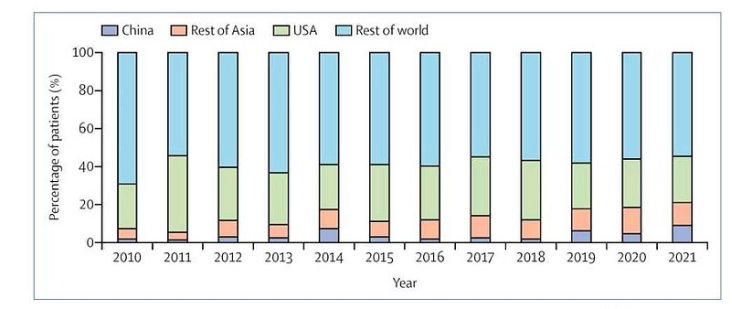

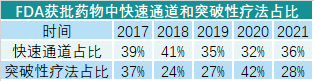
 版权声明/免责声明
版权声明/免责声明
推荐阅读

 点击这里,帮您找到理想的合作伙伴!
点击这里,帮您找到理想的合作伙伴!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权

重磅直播,精彩不容错过!
版权声明/免责声明推荐阅读
点击这里,帮您找到理想的合作伙伴!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来!
