如感兴趣,请联系我们(BD@drugtimes.cn)!
如果你是一个优秀的监管人员,在审批药物决定的过程中面临两种可能的错误:将无效的药物准入(Type I错误) 或将有效的药拒绝(Type II错误) ,哪个后果更严重一些?你可以参见“将无辜者入狱(Type I错误) 和“有罪者释放”(type II错误)。 在FDA的监管历史上,属于前者的错误越来越频繁地上演,其背后经常存在一个共同点:加速批准(AA,Accelerated Approval)。 加速审批可能是FDA政策中最具争议的一项,不断上演的“兴勃亡忽”的加速上市药物就印证了这一点。尤其是在肿瘤学领域,被撤回的加速审批俯仰皆是,暴露了加速批准过程中的诸多问题。最新发表在The Lancet Oncology上的一项研究用详实的数据和深入的分析,探讨了加速批准背后的是是非非。
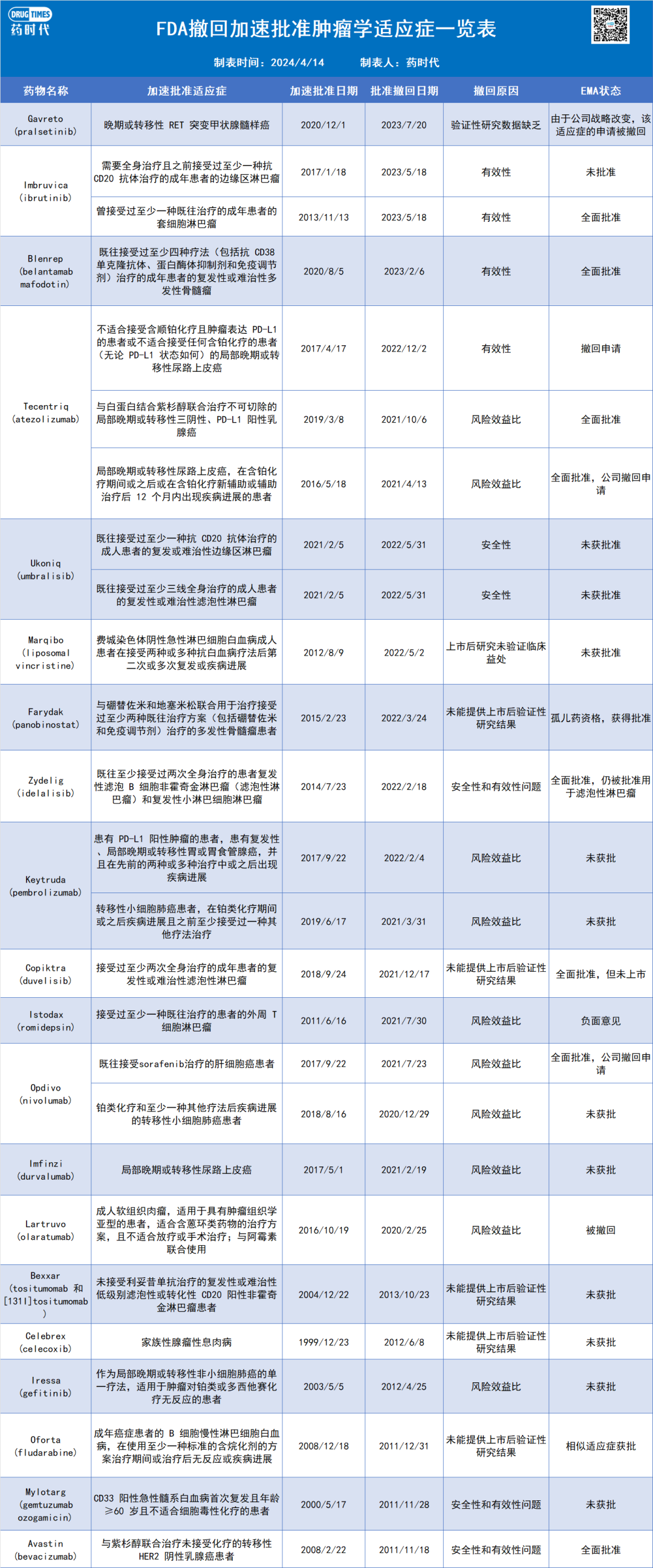
在过去的十年中, FDA和EMA批准了大量肿瘤学药物,包括很多突破性疗法和首创新药。常规的疗法监管审批过程中,人们已经习惯了随机对照试验 (RCT) 这条奉行的圭臬,而总体生存期和生活质量 (QOL, quality of life) 相应地成为评价药物临床意义的关键重点。然而FDA加速批准政策1992年的登堂入室,包括EMA有条件营销授权 (CMA,conditional marketing authorisation) 策略的出台,使得新产品“乱花渐欲迷人眼”的同时,也放大了监管审批过程中标准不一和透明度问题。“快进快出”新常态下,是一大批无效,甚至具有安全性问题的药物反常规地进入市场,为患者利益造成了很大的威胁。 针对在加速批准过程中可能的“大干快上”错误,FDA 在2021 年启动了 Project Confirm计划,期待通过提高加速批准的透明度,增强癌症药物的风险效益表现,来平衡加速批准过程中的受益与风险。一项研究显示,截至 2023 年 9 月 20 日,FDA撤回的23 种肿瘤学加速批准中,有9 种(39%) 仍然在欧盟拥有上市许可。2023 年 9 月,FDA 又撤回了3 个肿瘤学加速批准,使得被撤回的加速批准数量达到了26个(表1) ,这其中有20个(77%) 是在2020至2023年间被“批处理”撤回的,反应出FDA下达“罪己诏”纠错的决心和力度。在 FDA 撤销的 26 种适应症中,8 种仍获得 EMA 授权,15 种从未在欧洲获得批准,3 种已被 EMA 撤销。反过来看,有8 种肿瘤适应症通过 EMA 的 CMA 途径授权后被撤回,但从未获得 FDA 批准。监管机构之间决策不一致的部分原因,可能是制药公司提交的证据存在差异,这本身就导致了获取方面的差异。
表1. FDA撤回加速批准肿瘤学适应症一览表(信息来源:参考资料)
加速批准是是非非的争议离不开几个关键词:单臂试验和替代终点 (Surrogate endpoints)。 上文提及,总体生存期应该是常规批准肿瘤学奉行的终点圭臬,离开了这个“基本点”而批准的肿瘤学产品,很可能造成“差之毫厘,谬之千里”的结果。2008 年至 2012 年间,FDA 在缺少总体生存期或QOL 数据的情况下,批准了54款肿瘤学产品中的36个(67%) 。与之遥相呼应的是,2009 年至 2013 年间,EMA在没有支持生存证据的情况下批准了绝大多数肿瘤学药物。68 种药物中,只有 24 项 (35%) 批准拥有生存数据的背书,只有 7 种 (10%) 获得了QOL的支持数据。 临床试验设计也是加速批准的一个潜在问题。一项研究发现,2000年到2020年间,FDA批准的新型肿瘤学产品中,82%是在单一临床试验的基础上获批的,只有14%的主要终点是总体生存期。一半的新获批适应症(76/156) 没有随机对照试验证据。 其次,FDA和EMA在进行类似加速批准的监管决定过程中,似乎更关注安全性,或者说更偏向activity-risk平衡,优先于benefit-risk平衡。也就是说,如果有任何迹象表明某种药物对肿瘤表现出任何活性并且副作用很少,那么它就有可能获得许可。而国际监管机构越来越依赖 EMA 和 FDA 的决定。例如拉丁美洲和加勒比地区,各国明确接受依赖 EMA、FDA 和加拿大卫生部的营销授权。这种“唯马首是瞻”的依赖凸显了FDA两害相权取其轻的决策方针。但药物活性和患者利益之间还有一道临床益处验证的鸿沟。 为了填平这道鸿沟,FDA和EMA都要求加速批准的药物进行上市后的验证性研究,但很多申请者对此三心二意,甚至有故意拖延上市后研究的主观。 (84%) 仍然没有确凿证据表明它们有改善生存或生活质量的证据。 EMA 的 CMA 计划和 FDA 的加速批准之间也存在重要差异。大多数 FDA 的加速批准依赖于“可预测”的临床获益的终点,而 EMA 的 CMA则更依赖于风险收益比的评估。因此两大监管机构对于加速批准药物的上市后监管的侧重点也有所不同。FDA更关注产品的有效性。在某些情况下,对于风险收益比的评估会存在微妙的差异,这在一定程度上会导致了监管决策行为的前后不一致性。导致这种结果的部分原因可能是制药公司提交的证据(数量或质量) 存在差异。2023 年 3 月,FDA 出台了新的癌症药物指南草案,对加速批准途径进行了调整和改进。替代终点和单臂试验仍然是加速批准的可选性,指南对对照组也没有提出严格要求。 对于那些已经获得加速批准利益,而又不积极执行上市后验证性研究的开发商来说,他们的行为也越来越触动到了监管机构的底线。EMA 对 2009 年至 2013 年批准的肿瘤药物进行的系统评估显示,进入市场的 68 种药物中有 39 种 (57%) 没有证据表明它们可以延长生存期或改善生活质量。另外,23项疗法产生了相对于基线(现有疗法或安慰剂) 的优势,但实际上只有一小部分(11种) 真正具有临床意义。一旦癌症治疗在临床上实施,在实践中,即使长期数据没有显示生存或生活质量益处,也很难停止使用。 加速批准的肿瘤学产品的价格也是业界关注的焦点话题。很多药物的价格与其价值之间并不完全匹配。迄今为止,药物定价与临床获益量之间的关系仍然晦暗不明。
双盲随机对照设计是评估药物临床益处的最佳选择之一,它最大限度地降低了偏差(bias) 风险,并且明心见性地直指临床结果。从历史上看,在新疗法进入市场之前,进行至少两项 3 期随机对照试验应该成为黄金标准,可以显著降低“假阳性”(即统计学中的type I错误,即错误拒绝正确的H0 ,将无效的药物认定为有效)。 对于替代终点这个加速批准的“灵魂”,如果完全否定的话,加速批准可能也就失去了存在的意义,因此需要慎重对待。监管机构应该强调的,是更深入研究替代终点与总体生存期之间的关联,从而确认这些替代终点的有效性。 监管机构在某些申请者“加速批准、减速验证”的伎俩面前,也开始使用法律权杖进行反击。 例如2022 年 12 月签署FDORA《食品和药品综合改革法案》就赋予了FDA可以加速撤回批准而无需召开听证会的权力。同时,FDA也可以要求申请者在获得加速批准的同时,必须出示验证性研究“正在进行之中”的证据,否则可以拒绝监管批准。 展望未来,对于加速批准政策的优化将成为医药领域的重要议题。随着科技的不断进步和临床研究方法的改进,我们可以期待更精准、更高效的药物审批流程。未来的加速批准政策可能会更加注重数据科学和人工智能技术的运用,以加速药物和医疗器械的审批过程,并确保在加速的同时保持对安全性和有效性的严格监管。同时,未来还可能加强后续监管和实时数据监测机制,更及时地发现和处理药物或医疗器械的安全问题。通过不断地优化加速批准政策,有望为患者提供更快速、更安全、更有效的治疗选择,推动医学科技的进步,提升人类健康水平。 参考资料:
Koole, S. N. et al. Lessons learned from postmarketing withdrawals of expedited approvals for oncology drug indications. The Lancet Oncology. Volume 25, Issue 3, March 2024, Pages e126-e135
Kim , C. et al. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US Food and Drug Administration approvals. JAMA Intern Med, 175 (2015), pp. 1992-1994.
Davis, C. et al. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009–2013. BMJ, 359 (2017), Article j4530
Gloy, V . e t al. The evidence base of US Food and Drug Administration approvals of novel cancer therapies from 2000 to 2020. Int J Cancer, 152 (2023), pp. 2474-2484.
Gyawali, B. et al. The accelerated approval program for cancer drugs—finding the right balance
N Engl J Med, 389 (2023), pp. 968-971
其它公开资料
封面图来源:pixabay
版权声明/免责声明
本文为原创文章。
本文仅作信息交流之目的,不提供任何商用、医用、投资用建议。
文中图片、视频、字体、音乐等素材或为药时代购买的授权正版作品,或来自微信公共图片库,或取自公司官网/网络,部分素材根据CC0协议使用,版权归拥有者,药时代尽力注明来源。
如有任何问题,请与我们联系。
衷心感谢!
药时代官方网站:www.drugtimes.cn
联系方式:
电话:13651980212
微信:27674131
邮箱:contact@drugtimes.cn
本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权






 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 