



原系列文章共四篇,于2021年10月2日~5日在药时代上连载首发,受到广大朋友们的关注和好评。


糖尿病与胰岛素
在很长一段时间内,人们都认为糖尿病的发生,是由于肾脏损伤引起的。直到1776年,英格兰医生Matthew Dobson(1735-1784)发现,糖尿病患者的血清中也含有糖份,才让人们慢慢认识到“糖尿病是一种全身性慢性病变”的事实。
肠促胰素,Incretin
1932年,La Barre[1]首次提出“肠促胰素”这一概念,用于描述一种可降低血糖但不会引起胰腺外分泌的肠道上段黏膜提取物。1964~1967年,麦金太尔(McIntyre)[1]和埃尔里克(Elrick)[1]等人的独立研究发现,尽管静脉注射葡萄糖可达到比口服葡萄糖更高的血糖水平,但口服葡萄糖刺激胰岛素分泌反应的效应大于静脉注射葡萄糖,这种额外效应被称为“肠促胰素效应”。珀利(Perley)[1]等人进一步研究则证实,这种”肠促胰素效应”所产生的胰岛素占进食后胰岛素总量的60%左右。
1971年, Brown[1]等从小肠黏膜中分离出第一种肠促胰素,他们不仅发现了这种肠促胰素具有促进胰岛素分泌的作用,并发现这种作用具有葡萄糖依赖性,即只有当血糖升高时,才能促进胰岛素的分泌。也因此,这种肠促胰素被命名为葡萄糖依赖性促胰岛素分泌多肽(GIP)。
1985年,Schmidt等从肠黏膜中被分离提取出第二种肠促胰素,胰高血糖素样肽-1(GLP-1)。GLP-1由胰高血糖素原基因表达,在胰岛α细胞中,前激素转换酶2(PC2)将胰高血糖素原剪切为胰高血糖素,而在肠黏膜的L细胞中,前激素转换酶1(PC1)将胰高血糖素原剪切为GLP-1。GLP-1也具有葡萄糖依赖性,它和GIP一起共同产生肠促胰素效应。
1986年,Nauck[2]等人发现,2型糖尿病患者肠促胰素作用减退,提示肠促胰素系统异常可能是2型糖尿病的发病机制之一。Nauck等在对10例血糖控制不佳的2型糖尿病患者进行研究中发现,空腹状态下分别给予患者GLP-1或安慰剂,患者在输注GLP-1后,其胰岛素水平显著增加,胰高血糖素水平显著降低,空腹血糖水平在4小时后变为正常。而在血糖水平正常后,即使仍持续输注GLP-1,患者的胰岛素水平却不会继续升高,从而避免了低血糖的发生。这再次证明GLP-1具有葡萄糖浓度依赖性降糖作用,并免除了人们对现有糖尿病治疗药物及方案可能造成严重低血糖的担心。后续的临床研究发现, GIP的促胰岛素作用在2型糖尿病人几乎消失,可见病人中,GLP-1在肠促胰素效应中占据主导作用。
DPP4抑制剂
随着研究的深入,人们很快发现,天然GLP-1的一个很大的局限性就是,它在体内的半衰期特别短,分泌2-3分钟后就会被二肽基肽酶(DPP4)降解,即使外源性给予GLP-1,也同样很快会被DPP4降解。
基于此,人们设计了二肽基肽酶抑制剂,并最终成功用于临床。这对GLP-1来说,是一种“节流”的方式——降低GLP-1在体内的降解,从而达到降糖作用。于此同时,人们还在寻找“开源”的方式,期望找到一类“长效”GLP-1类似物,可以提高体内GLP-1(类似物)的浓度和作用时间。这种“开源”的方式,也许能够带来比“节流”方式更好的降糖作用。

Extendin-4及其类似物
20世纪70年代末和80年代初期,GIP和GLP-1等肽激素的发现,在科学界掀起了一股研究热潮,以了解这些新肽的作用机制、用途以及它们在体内受体的分布。其中,在曼哈顿岛北边的Veterans Affairs Medical Center(VAMC)/老兵事务医疗中心工作的罗莎琳·萨斯曼·耶洛(Rosalyn Sussman Yalow,1921-2011,1977年的诺贝尔生理学奖得主)则痴迷于在不同动物体内寻找新奇的激素,所使用的方法则是她自己的诺奖成就——Radio immuno assay/放射性免疫测定法。

By I, Blueag9, CC BY-SA 3.0,https://commons.wikimedia.org/w/index.php?curid=2292282
1992年,约翰·恩博士从一种名叫希拉毒蜥(Gila Monster)的毒液中,发现了两种成分,其中一种为首次发现,于是将其命名为Exendin-4。更重要的是,他还发现Exendin-4跟之前被发现的GLP-1的性质很像,在多种模型中,验证了Exendin-4能够刺激胰岛素的分泌。出于糖尿病医生的职业敏感性,他继续了对该多肽化合物的研究,并更惊喜地发现,Exendin-4的降解速度比GLP-1慢得多。
这本该是个令人振奋的发现,因为我们知道, GLP-1用于治疗糖尿病的困难,正是在于它在体内很快被DPP4降解(当时的麻省总院(MGH)也确有科学家正在研究用GLP-1治疗糖尿病)。然而,约翰·恩的发现并没有得到VAMC的重视,而原因竟是这一发现并不能解决老兵们的病痛。(也许,那些从战场退役下来的老兵们不容易患糖尿病吧?)
约翰·恩博士深信自己发现的价值,于是自掏腰包,花费了对一个普通研究员来说不菲的费用($8000),请律师申请了专利,并在1995年获得专利授权。
获得专利授权后,对约翰·恩博士来说,就是如何实现转化了,毕竟他自己没法把Exendin-4真正变成药物。于是,约翰·恩博士在1996年的American Diabetes Association Annual Meeting/美国糖尿病协会年会上,用海报(是的,他当时并没有获得发言机会)展示了Exendin-4在糖尿病小鼠中的“长效”作用。
Amylin病理学总监Andrew Young在会场看到了约翰·恩博士的展示,并非常清楚其价值,因为他们自己正在研发“长效”GLP-1类似物却一直没有成功,而这不正是他们一直在寻找的吗?真是踏破铁鞋无觅处,得来全不费工夫。Amylin第一时间买来了Exendin-4来进行测试,并立即开始与约翰·恩博士商量合作事宜。
糖尿病领域的巨头公司,Eli Lilly也看到了这篇poster,并与Amylin展开了竞争。然而,相比于Amylin公司所有关键决策人在一个会议室里的高效沟通决策模式,Eli Lilly则让约翰·恩博士和每个可能的决策人分别聊了半个小时,这让约翰·恩博士觉得更像是一次面试:I felt like it was a job interview for my compound.
就这样,约翰·恩博士在当年(1996)九月,与Amylin达成合作协议。Amylin非常看重这个产品,并在1998年完成了一期临床研究(Phase 1 study)。之后,尽管Amylin遭遇了与强生(J&J)在另一个糖尿病产品(Pramlintide)上的合作失败,几近破产,但仍保留了该项目的研发。

由于GLP-1类似物的降糖作用是葡萄糖依赖性的——避免了严重低血糖的风险,同时还被发现具有抑制食欲的作用——具有潜在的降体重效果,因此,尽管作为口服药物后的三线用药被批,很多患者却愿意直接使用Exenatide,使其成为Amylin收入的主要来源。
尽管Exenatide后来在2008年前后收到FDA关于急性胰腺炎的警告,并很快受到来自诺和诺德同类更优产品(me-better)利拉鲁肽的竞争(2010获批,每天注射一针,QD),使得其销售巅峰并未能持续多久,但它从希拉毒蜥毒液提取物(1992年约翰·恩博士的发现)到FIC糖尿病药物(2005年获批)这十三年的故事却可谓精彩纷呈,背后都是伟大科学家的付出与坚持,激励着我们一代代医药工作者。
面对竞争与挑战,Amylin在2009年和波士顿专业缓释制剂提供者Alkermes合作,很快研发出Exenatide的缓释微球制剂。尽管只是一个制剂的改变,其获批之路却并不顺利(Amylin原本非常自信地认为在2010年即可获批)。然而,FDA当年却给出了一封拒信,认为产品有质量缺陷,需要再提交一份Risk Evaluation and Mitigation Strategy(REMS)/风险评估和应对策略。
提交一个材料而已,赶紧准备一下,获批也还是指日可待的。然而,祸不单行的是,加拿大药监局通知FDA,认为早前他们要求Amylin在加拿大做的一个Exenatide对病人心电图影响的实验(n = 12),似乎有心血管风险!FDA立即通知Amylin,要求补交这个实验数据,作为批准长效Exenatide的申请材料。Amylin补交了实验数据,却是作为短效Exenatide的补充材料交的,认为这个不应该影响长效Exenatide的申请。
结果可想而知,FDA看了Amylin补交的关于短效Exenatide心电图实验数据,拒绝了Amylin关于长效Exenatide的上市申请。FDA要求Amylin重新开展一个临床实验,用新工厂生产的产品(之前临床实验用药是Alkermes提供的),证明长效Exenatide没有心血管风险。
所幸,长效Exenatide比短效Exenatide显示了更好的降糖效果,并且没有心血管风险,最终在2012年1月获批上市,成为第一款一周一次(QW)的GLP-1类产品。
这里还必须提一下,Sanofi(赛诺菲)后来通过开发Exendin-4的类似物,Lixisenatide/利西那肽,实现了每天一次给药(QD),并于2016年获得FDA批准(2013年获EMA批准)。
希拉毒蜥(Gila monster),一种不受欢迎的剧毒物种,因为给人类带来了Exendin-4,在CNN等报道中,成了“神奇的小家伙”。食量惊人的希拉毒蜥(一次可以吃下约为自身体重一半的饕餮大餐),也许正是凭借Exendin-4而避免受到“糖尿病”的困扰,实现了几乎所有人期望的“吃不胖”的梦想。
GLP-1类似物
如果要说糖尿病领域的龙头药企,当然非礼来和诺和诺德莫属。那么,当礼来通过于Amylin的合作,利用Exendin-4在GLP-1赛道拔得头筹时,诺和诺德是如何应对的呢?
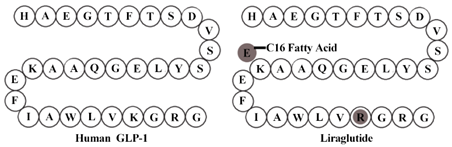
通过“alanine-scan”技术,已经了解到GLP-1的N端对与受体的结合非常重要,因此选择在靠近C端的位置引入脂肪酸。GLP-1中有两个Lys残基,他们将其中一个用Arg替代,从而避免了酰基化的选择性问题。在GLP-1与脂肪酸之间,他们还额外引入一个Glu,因为有报道显示,额外的羧基能够增强与白蛋白的结合[8]。这其实也很容易理解,脂肪酸酰化后,损失掉的一个羧基,正好由邻近的Glu上的羧基来补偿。进一步通过对脂肪酸长度的研究,他们发现更长的碳链可以带来更强的结合,并确定了C16和C18脂肪酸最优。
诺和诺德成功地不通过“抵抗DPP-4降解”的方式,找到了可以实现每天一次给药的Liraglutide,它在人体内的半衰期达到了13小时左右。而事实上,Liraglutide也是市场上唯一一个不“抵抗DPP-4降解”的GLP-1类似物。
Liraglutide不仅从结构上,对未来的GLP-1类似物开发指明了方向,同时在临床试验的方案设计上,也进行了开创性工作。GLP-1类似物有明显的胃肠道副作用,而临床上可以通过“titration”的方式,从低剂量慢慢增加到目标剂量,改善了患者的耐受性,从而可以提高临床剂量,以达到更好的药效。当然,这些也是诺和诺德的科学家们在临床实践过程中摸索发现的,而“titration”的方式很快成为该领域的标准方案。
Liraglutide(1.2/1.8 mg)于2009年通过EMA批准,2010年1月获FDA批准,用于治疗2型糖尿病。之后,Liraglutide(3.0 mg)于2014年通过FDA批准,用于成人肥胖患者;后又于2020年获FDA批准,用于12-17岁的肥胖患者,成为最畅销也最为安全的减肥药物。
诺和诺德Semaglutide的成功
夺得First-in-class的Amylin(后被AZ收购)自然不甘落后,前文已讲述,他们通过合作很快成功研发了缓释剂型,实现了每周一次的给药。然而,由于阴差阳错的原因,它直到2012年被FDA获批,比Liraglutide晚了整整2年。更为重要的是,它在后续的CVOT试验中,缓释剂型并未能展示出明确的心血管获益,终究棋差一招。Sanofi每天一次的Lixisenatide/利西那肽,更是不需多言,不仅降糖效果上不及前述产品,更是未见心血管获益。
GSK在2012年,通过收购人类基因组科学公司(HGS),闯入了GLP-1类似物的赛道。通过收购,他们获得了Albiglutide。诺和诺德的科学家,巧妙地利用脂肪酸链跟白蛋白的结合,成功研发了Liraglutide,HGS的科学家们则直接将GLP-1类似物(能够耐受DPP-4降解)跟白蛋白融合在了一起,实现了每周一次的给药。然而,III期临床数据显示,其降糖效果不及诺和诺德的Liraglutide。其他III期临床数据虽然显示它降糖效果优于Sitagliptin(DPP-4抑制剂)和Glimepiride(磺脲类降糖药),却有更严重的胃肠道副作用。尽管FDA最终还是在2014年批准了其上市,GSK还是在2018年宣布了其退市。也是在其退市的同年,Albiglutide的CVOT结果显示,它有明确的心血管获益。尽管如此,GSK还是毅然决然地退出了这一赛道的竞争,因为这个赛道除了诺和诺德这个强大的对手,另一个强大对手,Eli Lilly的度拉糖肽(Dulaglutide)也显示了强劲的竞争力。
Eli Lilly在GLP-1领域的布局已久,虽然前期的一些尝试以失败告终,但他们并没有放弃。即便通过与Amylin的合作,获得Exentide后,他们依然继续在GLP-1类似物领域耕耘。终于,他们也在长效GLP-1上迎来了突破。与GSK/HGS的策略类似,Eli Lilly将GLP-1类似物(能够耐受DPP-4降解)偶联到IgG4-Fc上,得到Dulaglutide,从而实现每周一次的给药。
在临床设计的竞争策略上,Eli Lilly选择了一条比较保守的策略,在临床III期设计与Liraglutide头对头对比的“非劣”试验,并最终取得了成功。Eli Lilly的Dulaglutide最终在2014年获得FDA批准,用于2型糖尿病患者,每周一次给药,剂量为0.75/1.5 mg。在后来的CVOT试验中,Eli Lilly也证明了Dulaglutide的心血管获益[12](虽然很勉强,但毕竟成功了)。
Dulaglutide的降糖效果非劣于每天一次的Liraglutide,降体重效果似乎不及,这也是他们没有申请减肥适应症的原因。在与Liraglutide的竞争中,Eli Lilly认为给药频次的降低,已经足够击败Liraglutide了,至少在糖尿病这个适应症上。事实证明他们是对的,但又没有完全对。因为他们真正的对手,并不是Liraglutide,而是诺和诺德。
诺和诺德深耕糖尿病领域多年,深知该领域的竞争之激烈,他们必然会在自己的每天给药一次的Liraglutide基础上,继续寻找其迭代产品。从马后炮的角度来看,诺和诺德在开发Liraglutide的时候,似乎就已经给未来留了后招——他们获得了脂肪酸链修饰的知识产权,利用脂肪酸链与白蛋白的高度结合,实现了QD给药,却在Liraglutide上,“放弃”引入针对DPP-4降解的改造。在成功推出Liraglutide后,诺和诺德的科学家们通过“捡起”针对DPP-4降解的改造,并再次优化了脂肪酸链(引入PEG基团,并在脂肪链端引入额外的一个羧基),于2017年成功推出了重磅产品——索马鲁肽(Semaglutide, 0.5/1 mg),用于2型糖尿病患者。
诺和诺德的目标很明确,他们要的不是me-better产品,而是真正的Best-in-class的产品——不仅需要给药频次降低至每周一次,而且需要有比Liraglutide更好的降糖降体重的药效。临床设计也服务于此,他们设计了对Liraglutide的头对头的优效试验,并取得了成功,“顺便”间接的击败了非劣于Liraglutide的Dulaglutide,可谓一箭双雕。他们还早早地完成了CVOT试验,证明了Semaglutide具有心血管获益[13]。
礼来很快也意识到竞争上的劣势,面对Semaglutide的强势出击,他们也很快进行了补救——在2018年开展了一个“higher dose Dulaglutide”的III期临床(NCT03495102),这个试验并不与任何其他药物进行比较,因此很快就在2019年完成,并在2020年获得了FDA的批准——3.0/4.5 mg剂量,用于之前低剂量不能维持药效的2型糖尿病患者。
诚然,在cross study的比较中,高剂量的Dulaglutide显示了优于Liraglutide,甚至Semaglutide的降糖效果,然而这终究不是一个严格的头对头临床比较。高剂量的Dulaglutide晚于Semaglutide三年后才获批,已经失去了先机。
2021年6月,高剂量的Semaglutide(2.4 mg)获得FDA批准,用于治疗肥胖患者,其68周降体重的效果达到12.7kg(12.4%,placebo adjusted),非常惊艳。基于Semagutide在临床的优异表现——糖尿病人群中最佳的降糖效果,肥胖人群中强大的降体重效果,明确的心血管获益以及肾脏获益,人们一度认为Semaglutide是GLP-1赛道,甚至2型糖尿病赛道的终极产品。未来还会有超越Semaglutide的产品吗?Eli Lilly在这个赛道还有机会吗?
Eli Lilly双重激动剂的反击
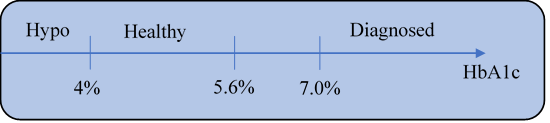
糖尿病的临床诊断,目前主要以糖化血红蛋白(HbA1c)含量划分(见上图)。糖尿病最重要的临床终点,也是糖化血红蛋白含量低于7%的人数比例。然而,这个指标并不代表血糖恢复正常。
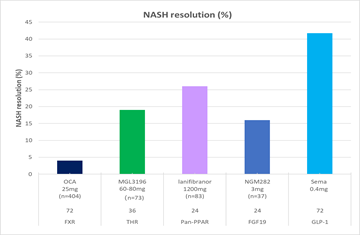
Eli Lilly在2018年完成的Dual GLP-1/GIP受体激动剂(Tirzepatide)在糖尿病人群中与Dulaglutide的头对头临床II期试验(NCT03131687),首次将“糖化血红蛋白含量低于5.7%的人数比例”加入到临床试验终点中,结果显示有43%的人在接受26周的治疗后,血糖恢复到正常人水平。在其他临床终点上,该化合物也展现出明显优于单独GLP-1类似物的降糖和降体重效果,可见,GIP成分在其中,必然起到了额外的作用。这与之前发现的“健康人对GIP敏感,而2型糖尿病人,似乎失去了对GIP的敏感性”[14]并不一致,推测可能是GIP和GLP-1之间存在协同作用。
基于近乎完美的临床II期结果,Eli Lilly迅速展开了多个III期临床试验,其中包括与Semaglutide的头对头试验(NCT03987919)。根据已经公开的数据,我们有理由相信,Tirzepatide相比Semaglutide有着更好的降糖和降体重效果,如果在不久的将来获批上市,它将会成为这个领域的重磅产品。Eli Lilly另辟蹊径,在Dual agonist赛道上扳回一城。
诺和诺德在这个方向自然也早有布局,甚至早于Eli Lilly,他们获得了最初由Marcadia Biotech开发的GLP-1/GIP活性都优于Tirzepetide的双激动剂NNC0090-2746(RG7697),并早在2015年,就完成了与Liraglutide的头对头的Phase IIa临床试验(NCT02205528)。然而,出人意料的是,这个化合物并没有显示出优效,甚至没有看到这样的趋势。可见,在双重激动剂方向上,还有很多不确定性存在,并不是简单的只要体外活性更好,就能带来更好的药效。
在GLP-1/GIP受体双重激动剂的赛道上,Eli Lilly和诺和诺德都有更多的布局;此外,有多个公司在GLP-1 plus的赛道布局,比如GLP-1/GCGR双重激动剂,GLP-1/GIP/GCGR三重激动剂等亦有相应的临床布局。在多重激动剂的赛道中最终谁能胜出,我们不能断言。但我们可以确信,正是在这种你追我赶的竞争中,病人将获得更好更方便的药物,而我们离治愈糖尿病的最终目标也将越来越近。
口服GLP-1R激动剂
然而,多肽类药物口服难有生物利用度,是一个公认的常识,主要原因就是它们在胃肠道被吸收前,很快就被胃液以及胃肠道中的各种酶降解了。是否能找到一种东西能够在GLP-1类似物被吸收前,保护它不被(快速)降解呢?
还真有人找到了这样的技术,诺和诺德也买到了这个技术。他们将Semaglutide与8-(2-羟基苯甲酰胺基)辛酸钠(SNAC)一起,通过SNAC在胃部形成局部的缓冲体系,中和了部分酸性,延缓了Semaglutide的降解;同时SNAC还能够促进胃壁细胞对Semaglutide的吸收,最终使得Semaglutide的生物利用度有了从0到1的突破——真正的0到1——生物利用度从0%提高到1%。也正是这一突破,实现了Semaglutide的口服给药,并于2019年获得FDA批准,7/14 mg,每天口服一次。口服与注射型Semaglutide7天总剂量的差别,几乎正好是生物利用度的差别。
然而,SNAC技术也并不完美,它至少有两个缺陷:1. 生物利用度在人和人之间的差别(variability)相比注射型更大,相应地带来治疗效果的差别;2. 糖尿病患者一般都患有其他疾病,相应地会服用其他药物,而SNAC有可能增加其他药物的吸收,从而带来潜在的风险。这也是为什么口服型Semaglutide对服药时间、饮食、其他用药的服用时间有诸多限制。正是这些限制,似乎偏离了我们所期望的“方便”。尽管如此,这仍然是口服GLP-1领域的一个里程碑。
怎样才能真正实现口服方便呢?小分子当然是不二选择。GLP-1及其类似物,都是做为GLP-1受体激动剂,与GLP-1受体结合后发挥作用的。那么,只要我们能找到小分子GLP-1受体激动剂,一切问题都会迎刃而解:我们不用担心多肽类药物的免疫原性,不用担心多肽类药物要避免的DPP-4降解,可以实现生物利用度的显著提高(相对于多肽)从而减少病人与病人之间的差别,未来甚至可以把它跟二甲双胍这样的一线用药做到一个片剂里面,等等。
但首先,我们必须要能够找到小分子GLP-1受体激动剂。
要让一个分子量只有几百的小分子去模拟一个长达30个氨基酸长度的多肽,谈何容易?小分子GLP-1受体激动剂,不是大家刚刚想到,刚刚意识到它可能带来的种种优点,而是几十年来,一直没有取得突破。
首先取得突破的是TransTech Pharma(后更名为vTv),他们发现了小分子GLP-1R激动剂——TTP054。这是一个部分激动剂,同时活性也比较弱,因此在临床上最高给到了800 mg的剂量(这已经都接近二甲双胍的高剂量了),每天一次给药,有一定的降糖效果,却不能降体重,因此在II期临床之后就停止开发了。
他们很快又推出了迭代产品——TTP273,这仍然是一个部分激动剂,但活性有所提高。临床剂量倒是降下来了,而为了达到更好的降糖效果,他们使用每天两次的给药方式,但临床结果却并不理想——降糖程度有限,更别提降体重了。
尽管TransTech Pharma的两代产品,并没有在临床取得成功,但他们的努力至少告诉我们两点:1. 我们是完全可以设计出小分子GLP-1R激动剂的;2. GLP-1R部分激动剂已经可以带来降血糖的作用了。
辉瑞(Pfizer)也早早开始进入在小分子GLP-1R激动剂领域,所使用的寻找苗头化合物的方法就是大公司擅长的高通量筛选(HTS)——因为他们拥有自己独特的小分子化合物库。我们完全有理由相信,许多家跨国药企都早就开始使用HTS的方法在寻找小分子GLP-1R激动剂。
这注定是一个艰难的工作,小分子替代比它大得多的多肽,挑战实在太大了。直到Pfizer的科学家们在筛选的Assay上做了一个小小的,却非常重要的改变。而这一改变的逻辑并不复杂——如果小分子没有在生物Assay中体现出阳性信号,不是小分子的活性不够好,就是Assay不够敏感。我们不能控制化合物库里小分子的活性,还不能提高一下Assay的敏感性吗?对GPCR(GLP-1R是Class-B家族GPCR的一员)来说,增加敏感性并不难,只要在Assay里加入GPCR的正向别构调节剂(PAM)即可,而GLP-1R的PAM已经有很多报道了。
正是生物筛选Assay上这一小小的改变,让Pfizer通过HTS的方法找到了hit,并最终找到一个小分子GLP-1R的完全激动剂——PF-06882961[15]。该化合物已经完成了多项临床II期试验,在28天的MAD试验(NCT03538743)中,该化合物已经显示了明确的降糖降体重效果,未来可期。
PF-06882961在临床上需要每天两次口服给药,因此Pfizer很快也推出了迭代产品——PF-07081532,并已经完成了临床I期试验(NCT04305587)。
Chugai公司也找到了自己的小分子GLP-1R激动剂,并在2018年以50M首付款的交易将其临床前小分子GLP-1R激动剂项目转让给Eli Lilly[16]。Eli Lilly认为该化合物是一个条件依赖的完全/部分激动剂,有可能在临床上带来更好的耐受性[17]。目前该化合物已经完成了临床I期试验(NCT04426474),并进入临床II期阶段。

GLP-1在其他适应症的应用
GLP-1R激动剂临床试验的开展以及上市后的使用,提供了越来越多的实际使用数据,我们可以看到GLP-1R激动剂除了降糖降体重外,还能让糖尿病人有心血管获益[18],对肾脏也有一定的保护作用[19]。此外,GLP-1R还具有许多其他生物学特性及功能,可通过作用于中枢增强学习和记忆功能,因此针对于阿兹海默症的临床研究也正在进行(NCT04777409,NCT04777396)。

本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 