



临床成功的ADCs的设计不仅取决于有效载荷的效力及其附着点、连接子的稳定性和有效的药物释放,而且还取决于抗体和生物偶联技术的选择。在过去10年中,所有经FDA批准的ADC都是以ADC的异构混合物的形式存在,单抗的不同位置上附着着不同数量的药物。偶联位点对ADC稳定性及其药代动力学具有显著的影响,高DAR(药物抗体比)常常导致血浆清除迅速,而低DAR的ADC则表现出的活性较弱。在ADC的药物中,裸单抗的存在是一种有效的竞争抑制剂。因此,在过去十年中,人们开发了一大批新的偶联策略,目的是控制小分子药物的位置和数量,同时保持结构完整性和同质性。
单克隆抗体的天然结构为生物偶联提供了多种可能性,基于化学的、特异性的天然(非工程)抗体偶联具有一些优点。它可以避免抗体特定位点突变的复杂性,以及在细胞培养的放大和优化方面可能面临的挑战。
偶联位点根据抗体序列,赖氨酸、组氨酸、酪氨酸和半胱氨酸等内源性氨基酸在二硫键间的连接位点非常具有吸引力。所有经FDA批准的ADC,直到2021年,都利用这些内源性氨基酸进行偶联。然而,抗体支架还包含聚糖,这是在单克隆抗体生产过程中,FC区域的翻译后修饰所导致的。一些研究报告了糖工程化的新策略,这似乎是一种有趣的生物偶联替代方法。
与内源性氨基酸的偶联
最常见的偶联方法之一是利用抗体的赖氨酸残基,氨基酸亲核NH2基团与利克有效载荷上亲电的N-羟基琥珀酰亚胺(NHS)基团发生反应。尽管反应简单,但可利用赖氨酸残基的高丰度导致了许多ADC在随机分布下的不均匀混合物的形成。DAR受药物/抗体化学计量比的控制,该方法得到广泛应用,包括已获批的ADC,如Besponsa, Mylotarg, 和Kadcyla。
最近,同样也报道了对赖氨酸位点和残基的特异性修饰。通过计算机辅助设计,磺酰丙烯酸酯被用作中间试剂,用于在天然蛋白质序列上对单个赖氨酸残基进行修饰。
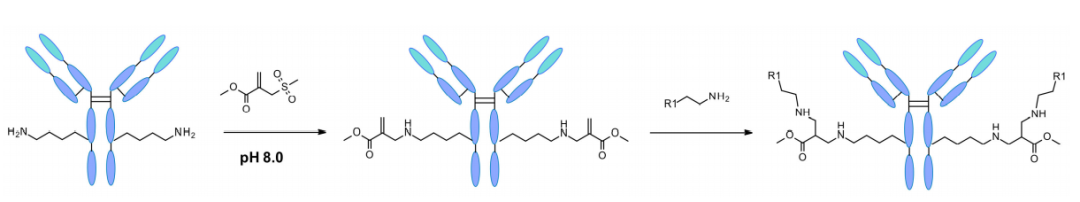
反应的区域选择性归咎于磺酰丙烯酸酯的设计以及每个赖氨酸周围独特的局部微环境。通过计算预测,pKa最低的赖氨酸容易以位点特异性的方式在弱碱性pH下优先反应。即使在其他亲核残基如半胱氨酸存在的情况下也观察到了这种反应。该技术已应用于5种不同的蛋白质和曲妥珠单抗,在偶联后均保留了原有的二级结构和蛋白质功能。
2018年,Rai等人报告了另一种利用“化学关键蛋白”的可逆分子间反应进行的位点特异性修饰。该试剂携带多种官能团,这些官能团在所有可获得的赖氨酸残基上可逆地形成亚胺部分。然后,关键蛋白通过试剂中的环氧化物与近端组氨酸残基反应。因此,在生理条件下,关键蛋白从赖氨酸中分离,醛被再生,从而能够通过肟结合标记抗体。
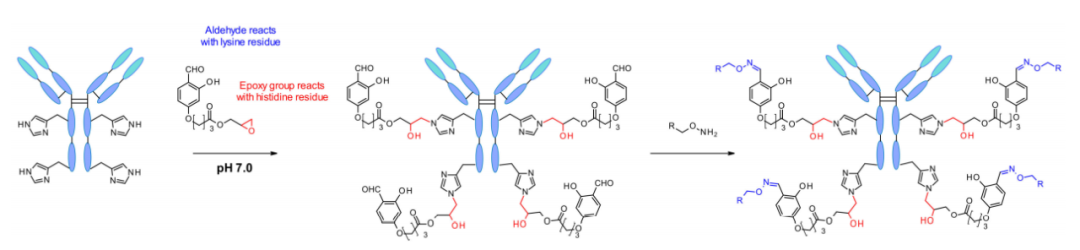
这种关键蛋白的定向修饰技术后来发展成单赖氨酸残基标记技术,即使在存在N-末端胺的情况下也具有毋庸置疑的选择性。方法的成功依赖于Fk1-间隔子-Fk2试剂。
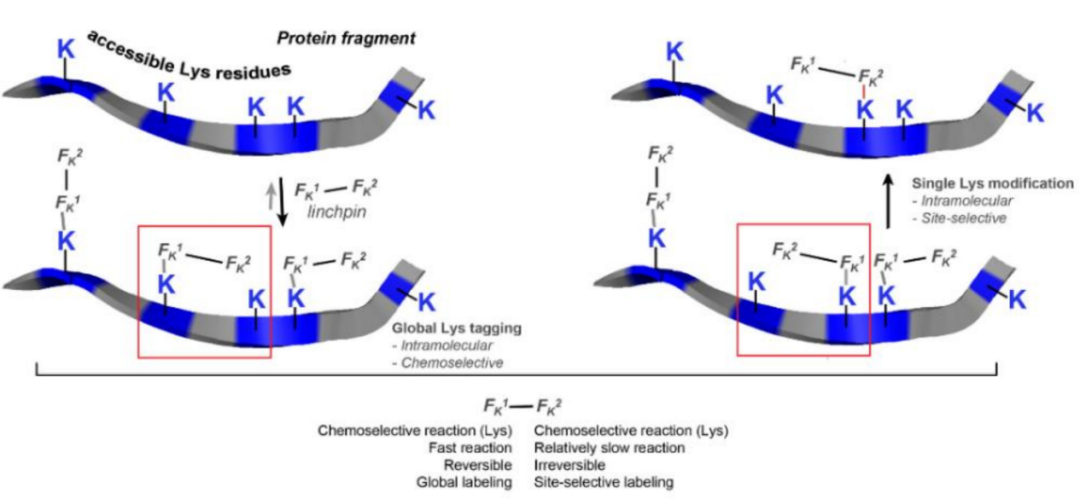
Fk1官能团与赖氨酸可逆反应,调节Fk2近端赖氨酸部分的微环境。然后通过酰胺键在Fk2处的赖氨酸残基(K169和K395)进行偶联,间隔子的设计调节偶联的位置。该方法已成功应用于ADC(trastuzumab-emtansine)的合成,证明其细胞活性与已获批准的Kadcyla相当。
Merlul等人最近报道了一种不同的结合策略,有效地靶向天然抗体上的组氨酸残基。他们引入了一种基于阳离子有机金属铂(II)的连接子,[乙二胺铂(II)]2+,图中表示为Lx。
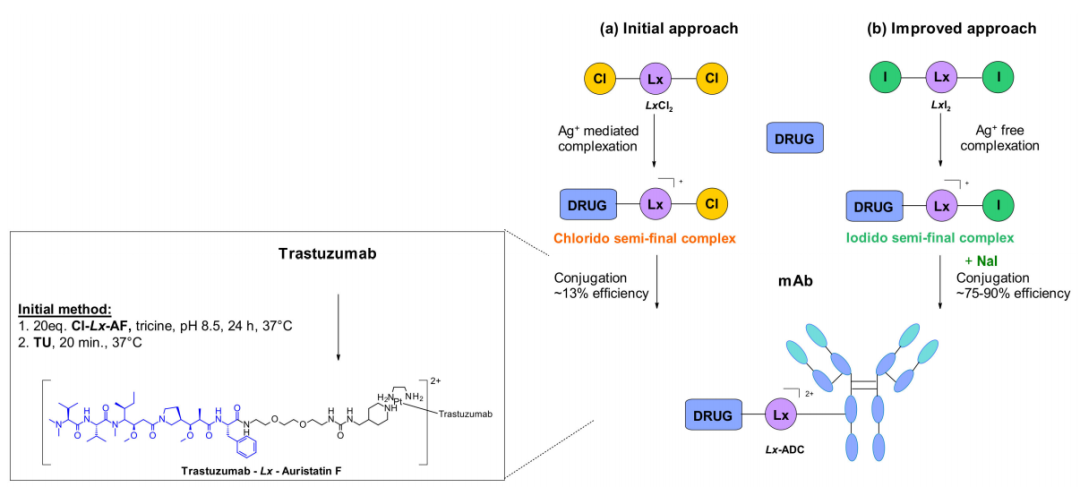
这种技术基于络合和偶联两个步骤。氮杂环配体如哌啶与Lx配位形成络合物前体,稳定的中间体包含有效载荷和配体上的一个氯离子。该复合物含有带正电荷的Pt(II)中心,这提高了连接子和有效载荷复合物的水溶性并最小化抗体聚集,该方法还扩展到类似的碘络合物。在最近的一份报告中,碘化钠的使用被证明可以显著提高该技术的偶联产率和选择性。Cl-Lx-药物载荷络合物上残留的氯配基与碘化物的交换生成更具活性的I-Lx-药物载荷,从而获得更高的偶联产率。这项技术已被应用于ADC药物的大规模生产。
二硫化物重桥接策略
IgG抗体包含四个链间二硫键,两个连接轻链和重链,两个位于连接两条重链的铰链区,它们维持着单克隆抗体的完整性。另一个经典的生物偶联途径探索了这些半胱氨酸作为有效载荷连接点的作用。四个二硫键的还原通常会产生八个巯基,它们能够与马来酰亚胺的连接子反应,从而产生DAR为8的ADC。
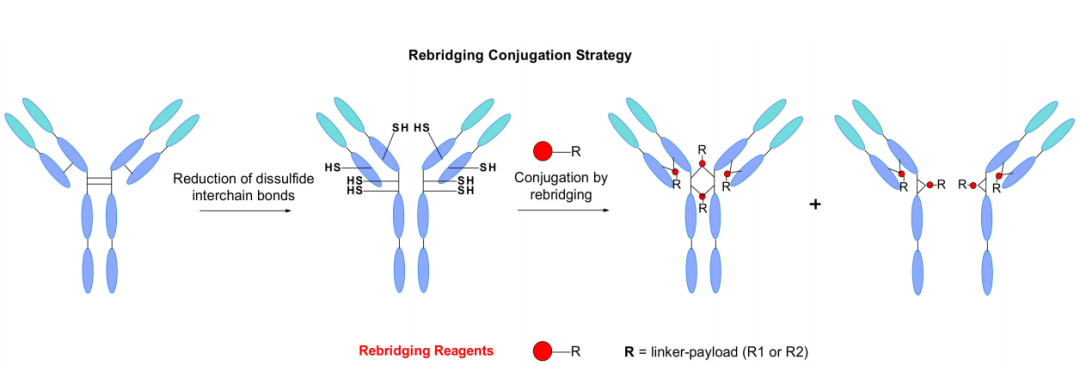
Doronina及其同事报告了嵌合抗CD30单克隆抗体偶联MMAE,DAR=8的 ADC实例。与经典的赖氨酸偶联相比,这种有效载荷加载方式得到了更好的控制。然而,据报道,较高的药物负荷会增加聚集的风险,从而导致高血浆清除率,并降低体内疗效。
Badescu和al在2014年报告了一种新的位点特异性重桥接偶联策略,他们是第一个证明新的双砜(bis-sulfone)能够烷基化来自抗体和抗体片段中还原二硫键的两个巯基,对抗原结合的影响最小。后来,Wang和al描述了一种新的水溶性烯丙砜(allyl sulfone),该试剂在没有原位活化的情况下提高了反应活性。它表现出高稳定性、高水溶性和位点特异性。
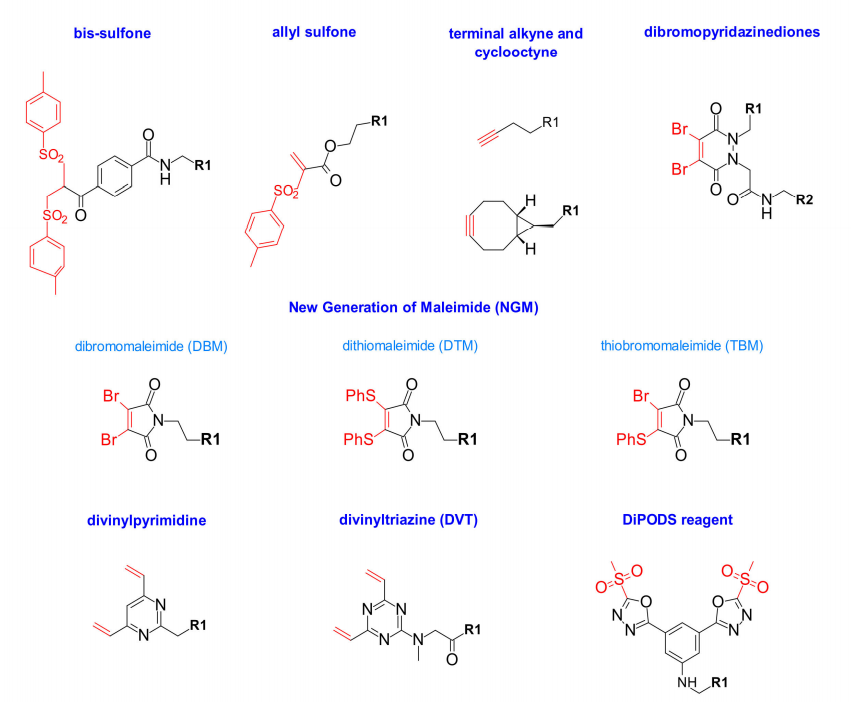
此外,还有巯基炔与末端炔烃和环辛炔生物偶联的再桥接技术,其进一步发展出新一代马来酰亚胺,如二溴-(DBM)和二硫代马来酰亚胺(DTM),用于位点特异性偶联。这些马来酰亚胺类似物在第3位和第4位含有良好的脱离基团,从而实现快速、高效和高产率的偶联。最近报道了结合二溴和二硫代马来酰亚胺性质的杂化硫代溴马来酰亚胺(TBM),这种TBM试剂结合更快,显示出更高的DAR=4的百分比,这可能是由于溴减少了空间位阻。
2015年,Chudasama等人引入了一类新的重桥接试剂,二溴吡啶二酮(dibromopyridazinediones)。他们证明了它能有效地插入到二硫键中,得到的结构即使在高温下也表现出了极好的水解稳定性。然而,随着还原步骤上的温度升高,也观察到不均一性,这种结构也允许选择性地引入不同的功能基团。
二乙烯基嘧啶(Divinylpyrimidine)是另一种有效的重桥接试剂,能够产生稳定的DAR=4的ADC。Spring等人研究了乙烯基杂芳基支架对半胱氨酸再桥接的作用,他们认为用嘧啶取代吡啶可以使杂芳环成为更好的电子受体,从而提高交联效率。他们的工作扩展到二乙烯基三嗪,在高温下,重桥接显示出更高的效率。
为了避免与经典马来酰亚胺偶联相关的体内不稳定性的缺点,Barbas等人研究了甲基磺酰基苯基恶二唑,该试剂对半胱氨酸具有特异性反应。与血浆中的半胱氨酸-马来酰亚胺偶联物相比,他们的稳定性更高。受此启发,Zeglis设计了DiPODS试剂,该试剂含有两个通过苯基连接的恶二唑基甲基砜部分, DiPODS以重桥接的方式与两个硫酸根形成共价键。与马来酰亚胺偶联相比,以这种方式偶联具有优越的体外稳定性和体内性能。
聚糖偶联
由于IgG是一种糖蛋白,它在Fc片段每个重链的CH2结构域N297位置包含一个N-聚糖,这种糖基化可以作为连接有效载荷的附着点。多糖与Fab区域间远距离定位降低了在偶联后损害抗体的抗原结合能力的风险,此外,与抗体的肽链相比,它们的化学组成不同,允许位点特异性修饰,使它们成为合适的偶联位点。
聚糖生物偶联可根据用于靶向碳水化合物的技术来区分:包括聚糖代谢工程化、聚糖氧化后的糖转移酶处理、内糖苷酶和转移酶处理后的酮或叠氮化物标记。
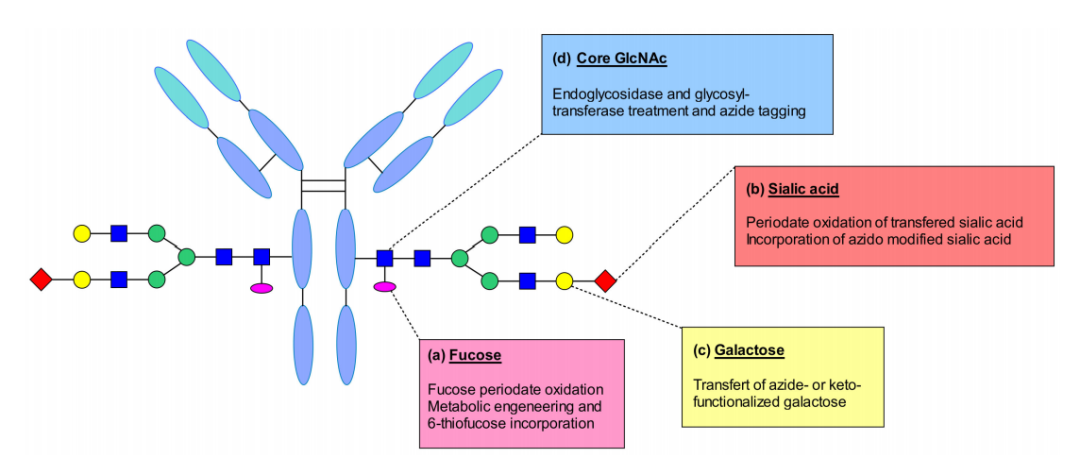
Neri等人报道了在IgG抗体的N-糖基化位点处岩藻糖的位点特异性修饰。这种糖含有一个顺式二醇部分,适合选择性氧化。他们用偏高碘酸钠氧化岩藻糖残基,生成一个能够与含联氨的连接子反应的醛基,这样,抗体通过腙键与药物相连。
Senter及其同事向细胞培养基中添加硫基类似物,通过代谢将6-硫代岩藻糖带入抗体修饰。他们认为,取代是通过劫持岩藻糖基化途径来完成的,这样就引入了化学位点来实现位点特异性结合。与经典半胱氨酸偶联物相比,这种方法显著降低了异质性水平,并产生具有更可预测的药动学和药效学特性的偶联物。
重组IgG中很少含有唾液酸,然而,已经证明,利用半乳糖基和唾液酸转移酶可以酶法改造甘氨酸。通过酶反应添加半乳糖以获得G2聚糖,然后添加末端唾液酸。这种修饰通过高碘酸氧化生成醛基,可以偶联带羟胺基团的连接子-有效载荷。所得的偶联物具有较高的靶向选择性,体内抗肿瘤活性良好。高碘酸还可氧化蛋氨酸等敏感氨基酸,影响与FcRn的结合。
除这些偶联策略外,半乳糖残基也可以作为修饰位点。多项研究报告了通过使用突变的β- 1,4-半乳糖转移酶,将半乳糖替换为一种含酮或叠氮官能团的半乳糖,这种具有双正交官能团的半乳糖衍生物为高效偶联开辟了途径。这些技术已被开发用于成像和抗癌应用。
从化脓性链球菌中发现的内糖苷酶EndoS和EndoS2,这些酶能够水解IgG的N-聚糖,从而使水解后的残基成为生物偶联的有效位点。这种方法有助于使单抗的聚糖结构均匀化,同时它也适用于任何IgG亚型。此类方法应用于trastuzumab-maytansine,制备出具有良好体外和体内药效的糖偶联ADC。
生物正交化学和蛋白质工程领域的进展有助于产生更均匀的ADC。尽管在天然单抗上有许多可用的附着方法可供选择,但在工程化抗体上的位点特异性生物偶联能够更有效地控制DAR,并且避免改变与抗原结合的亲和力。这样,在某些位置加入天然或非天然氨基酸,得到具有优良药代动力学和药效学特征的同质产品。
酶法
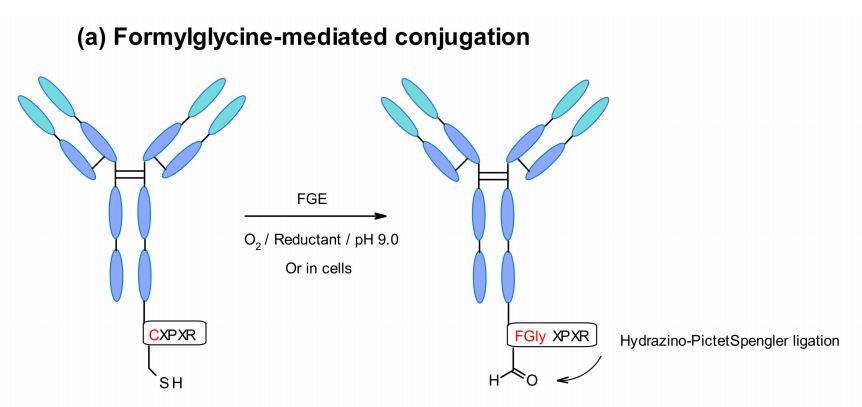
有效载荷的附着可以通过在抗体序列中插入特定的氨基酸标签以非常有选择性的方式实现。这些标签被特定的酶所识别,例如甲酰甘氨酸生成酶(FGE)、微生物谷氨酰胺转胺酶(MTG)、转肽酶或酪氨酸酶,从而能够执行位点特异性偶联。
Aaron等人探索了一种新的利用醛标记蛋白质的位点特异性偶联。该技术利用了基因编码的五肽序列(Cys-X-Pro-X-Arg),其中半胱氨酸残基被FGE识别,并在细胞中蛋白质表达期间被共翻译氧化为甲酰甘氨酸。这样,工程化抗体通过HIPS(hydrazino-Pictet–Spengler)化学方法与醛特异性连接子选择性偶联。
微生物转谷氨酰胺酶(MTGase)策略也经常被开发用于定位特异性偶联。MTGase催化在脱糖抗体295位置的谷氨酰胺侧链与底物的伯胺之间形成肽键。与其他酶策略相比,MTG是一种灵活的技术,不需要肽供体来实现偶联。只要酰基受体含有一种伯胺,就没有结构限制。
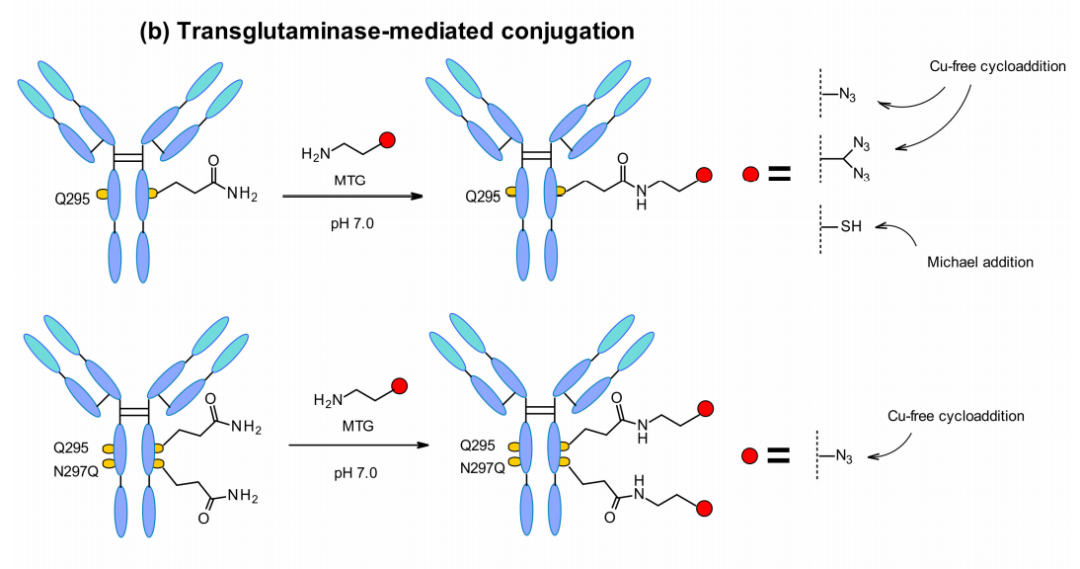
谷氨酰胺残基自然存在于单抗的每个重链的Fc区域。在295位去糖基化后,谷氨酰胺残基通过MTGase介导的反应偶联,可以产生均一的DAR=2的ADC。为了提高效率,可以偶联带支链的连接子,从而使DAR翻倍,297位的天冬酰胺突变为谷氨酰胺也可增加DAR。
NBE Therapeutics开发了基于S金黄色葡萄球菌转肽酶A介导的偶联。他们的策略利用转肽酶A(SrtA),在LPXTG(X=任何氨基酸)五肽的基序中切割苏氨酸和甘氨酸残基之间的酰胺键。然后,它催化甘氨酸相关的有效载荷与新生成的C-末端的偶联,在生理温度和pH下生成肽键。
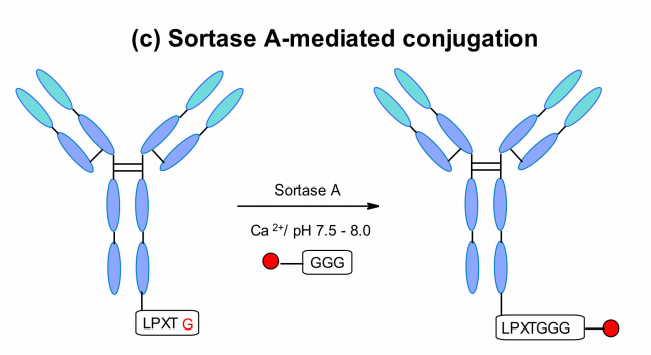
该方法应用于不同抗体,如抗CD30和抗Her2,并使用含有5甘氨酸标记的连接子偶联maytansine和MMAE,两种ADC均显示出与经典偶联相似的体外细胞杀伤活性。酶法产生的trastuzumab-maytansine在体内试验中完全匹配Kadcyla。
在另一个例子中,利用转肽酶法生成了高效蒽环素毒素衍生物PNU-159682的ADC。有趣的是,通过这项技术,偶联效率甚至高于Adcetris和Kadcyla类似物。此外,所制备的PNU-159682 ADC具有较高的体外和体内稳定性,并且显示出的效力超过了含有微管蛋白靶向有效载荷的ADC。
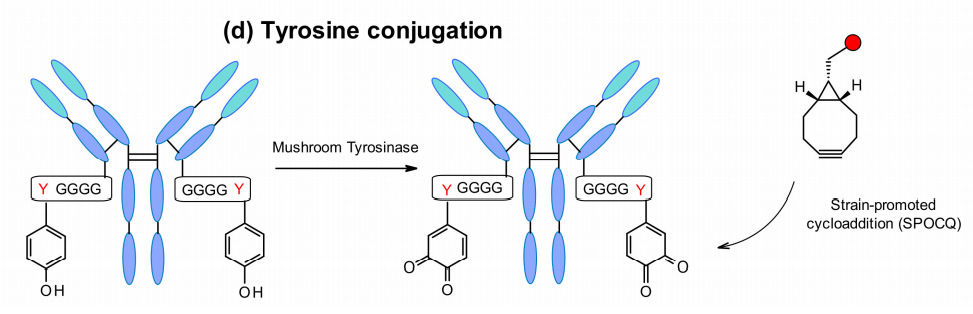
另一个新兴的新方法是通过酪氨酸标签进行位点特异性抗体标记,酪氨酸标签与单克隆抗体轻链的C末端基因融合。考虑到位点可及性,Bruins及其同事使用了一种工程化的四甘氨酰酪氨酸残基作为标记,它为偶联提供了一个容易触及的位点。酪氨酸酶将酪氨酸氧化成1,2-醌,从而允许与各种双环[6.1.0]壬炔(BCN)衍生物的环加成反应。这种方法可以与含有BCN连接子的MMAE有效地偶联。
半胱氨酸工程:硫单抗技术
随机半胱氨酸偶联和重桥接是利用抗体结构内天然存在的半胱氨酸残基的技术。然而,随机半胱氨酸方法的异质性以及重桥接策略中的单抗片段化需要在ADC合成中加以考虑,特别是当疏水性药物被偶联时。
与它们不同的是,硫单抗技术通过利用不涉及结构二硫键的工程化反应性半胱氨酸,在抗体上实现所需位点的选择性和均匀修饰。一般来说,半胱氨酸突变的设计是为了促进细胞毒性有效载荷偶联的同时,保持单克隆抗体的稳定性、亲和力和最小化ADC聚集。为了确定突变的最佳位置,通常采用几种技术,包括计算建模、模型系统筛选和高通量扫描。
Junutula等人首先报道了一种硫单抗策略,用工程化半胱氨酸残基取代了抗MUC16抗体重链114位的丙氨酸(HC-A114),工程化位置内的反应性硫醇能够与马来酰亚胺负载的连接子反应。合成的抗MUC16 ADC在异种移植小鼠模型中表现出效力,在大鼠和食蟹猴中表现出高剂量耐受性,这个发现建立了硫单抗偶联策略的一般性方法。
此外,琥珀酰亚胺连接在胞浆内可以经历两个平行反应:反向Michael反应导致连接子-有效载荷的损失,以及琥珀酰亚胺的水解,这两种反应都对体内ADC活性有显著影响。为了提高稳定性,Lyon和合作者设计了一个与马来酰亚胺相邻的碱性氨基整合进来的连接子。在连接子中加入二氨基丙酸(DPR)促进了硫琥珀酰亚胺在中性pH和室温下的快速定量水解,这样,非特异性的去偶联作用被阻止,从而提高了体内的稳定性。除了常用的马来酰亚胺外,还探索了不同的半胱氨酸反应剂,如碘乙酰胺、溴甲酰胺、羰基丙烯酸酯,N-烷基乙烯基吡啶盐。
除了硫单抗技术外,非标准氨基酸(ncAA)的加入为位点特异性偶联提供了另一种可能性。该技术使用含有独特化学结构的氨基酸,从而能够以化学选择性的方式引入连接子-有效载荷复合物。该技术需要对抗体序列重组,利用与宿主细胞内所有内源性tRNAs和合成酶正交的tRNA和氨基酰tRNA合成酶(aaRS),用于响应未赋值密码子将ncAA带入蛋白质。通常,ncAA在发酵过程中被添加到培养基中。选择非天然氨基酸是很重要的,因为它们可能激发免疫原性。常用的ncAA是具有独特基团的天然氨基酸的类似物,如酮、叠氮、环丙烯或二烯。
已有研究将对乙酰苯丙氨酸(pAcF)成功地整合入抗CXCR4 抗体中。有效载荷Auristin通过肟连接与抗体有效偶联,从而生成化学均一的ADC。该ADC在小鼠体内表现出良好的体外活性和完全清除肺肿瘤的作用。
由于肟连接所需的酸性条件和ADC缓慢释放的动力学,另一种选择是加入含ncAA的叠氮化物。广泛应用的对叠氮哌苯胺(pAzF)可在生理条件下快速进行CuAAC或SPAAC反应,利用这种策略成功地在抗CD74抗体上偶联糖皮质激素有效载荷。除了pAcF技术外,还成功地将含叠氮的赖氨酸类似物(AzK)带入到抗体中,以产生具有Auristin、PBD二聚体或微管蛋白有效载荷的位点特异性ADC。
此外,赖氨酸的环丙烯衍生物(CypK)以及自然发生的非典型氨基酸,如硒代半胱氨酸(Sec)都成功地整合进入抗体中。所产生的ADC表现出良好的稳定性、选择性以及体外和体内活性。
在过去的几年里,ADC的结构优化和机制扩展方面取得了许多进展。新的偶联技术已经被开发出来,以获得对肿瘤的更高选择性。这些偶联技术使ADC具有更好的稳定性、选择性以及体外和体内活性。这些新型偶联技术的初步数据令人鼓舞,未来将极大地促进ADC药物的迅猛发展。
参考文献:
1.The Chemistry Behind ADCs. Pharmaceuticals (Basel). 2021 May; 14(5): 442.
版权声明/免责声明
推荐阅读
 点击这里,帮您找到理想的合作伙伴!
点击这里,帮您找到理想的合作伙伴!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 