相比传统的随机对照的金标准试验模式,以早期单臂研究(简称SAT)即可申请上市政策的出现极大缩短了药物研发上市的周期。
据统计,相比常规申请上市进度,单臂上市可节省新药中位近2年的临床获批时间。目前国内外对单臂上市的接受度和成绩如何?我们来看一下数据。
我国自去年CDE颁布《单臂试验支持上市的抗肿瘤药进入关键试验前临床方面沟通交流技术指导原则》后,单臂上市的比例及成功药物逐渐增多。
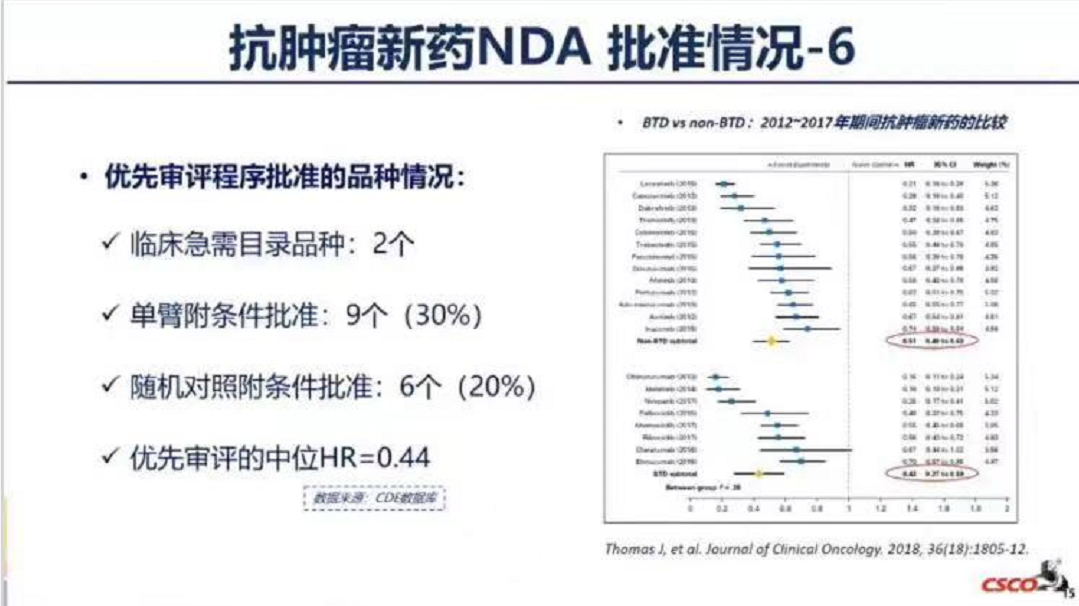
在上月CSCO大会CDE审评专场中,根据杨志敏部长的《抗肿瘤新药附条件批准的审评考量》中的数据统计显示,本年度截至2021年9月10日,国内共获批上市45种新药,有30种纳入优先审评,而其中9个(30%)通过单臂附条件获批上市!
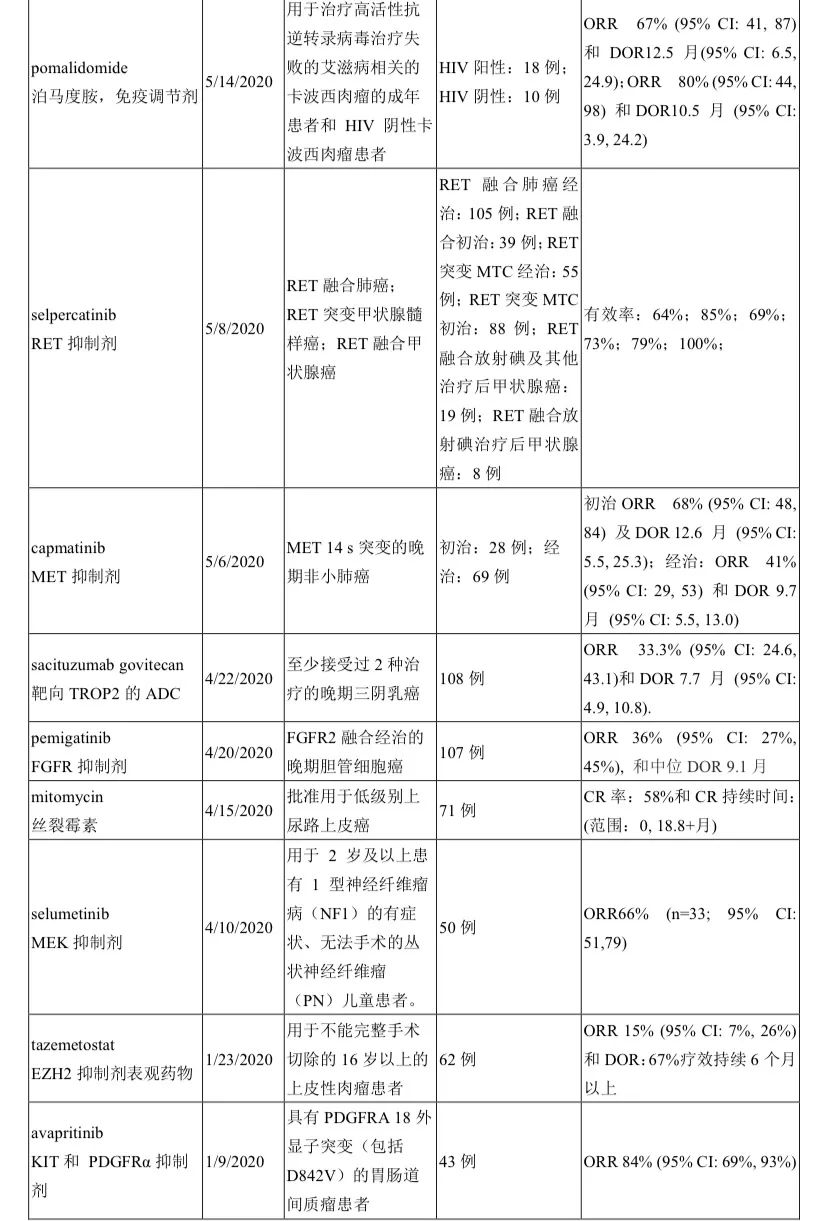
包括特瑞普利单抗(鼻咽癌)、伏美替尼(EGFR T790M肺癌)、普拉曲沙(RET肺癌)、阿伐替尼(PDGFRA18突变GIST)、特瑞普利单抗(尿路上皮癌)、卡瑞利珠单抗(鼻咽癌)、帕米帕利(gBRCA的卵巢癌)、维迪西妥单抗(HER2阳性胃癌)、赛沃替尼(MET14突变肺癌)。具体上市审批见以下截图。
从国内单臂上市的药物来看,靶向和免疫是主力军,ADC也逐渐崭露头角。
靶向药物,由于对覆盖靶点的高效性和安全性,是单臂注册上市的主要类型,1类新药+明确靶点+高疗效,基本可以促成一个靶向药物的快速上市,是未来国内外靶向药物首发上市的主导模式。
而PD1免疫方面,以国外还未挖掘出的鼻咽癌的适应症成功获批了国内的两大PD1,可见新适应症的挖掘是在多种同类竞品中脱颖而出的关键。而纯创新类新药如RC48(维迪西妥单抗)配上可看的疗效,单臂上市的成功率很高。
国内对于单臂上市的倡导和开放时间尚短,只能说初展模式,但势头强劲,快速熟悉单臂上市的要点和走位,从药物属性到开发策略到具体设计,充分考量!
国际FDA:超1/3新药单臂上市,全癌肿覆盖,创新+高效是核心!
以上国内注册数据我们仅看到了国内成功产品的早期特征,那对于单臂上市盛行多年的FDA数据,我们似乎能看到更多单臂注册上市的发展特质和突破方向!
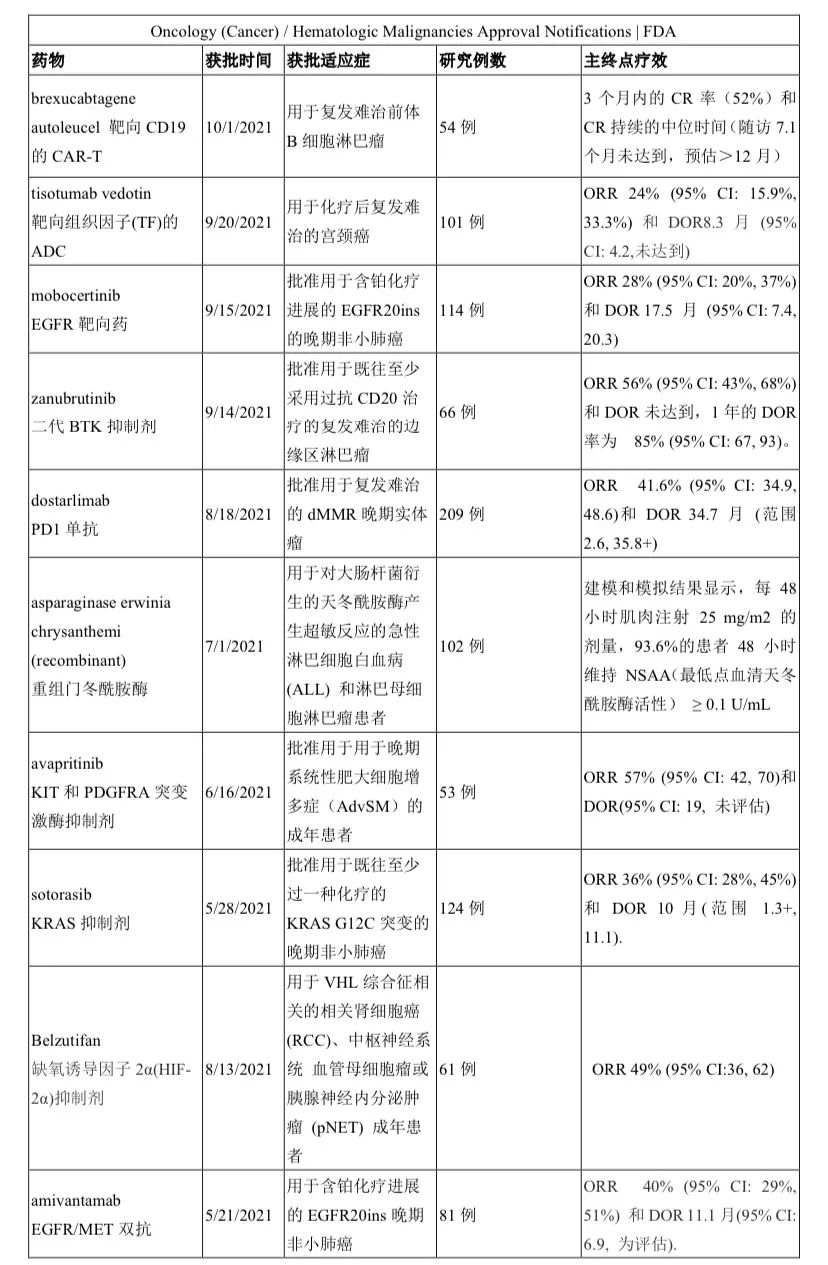
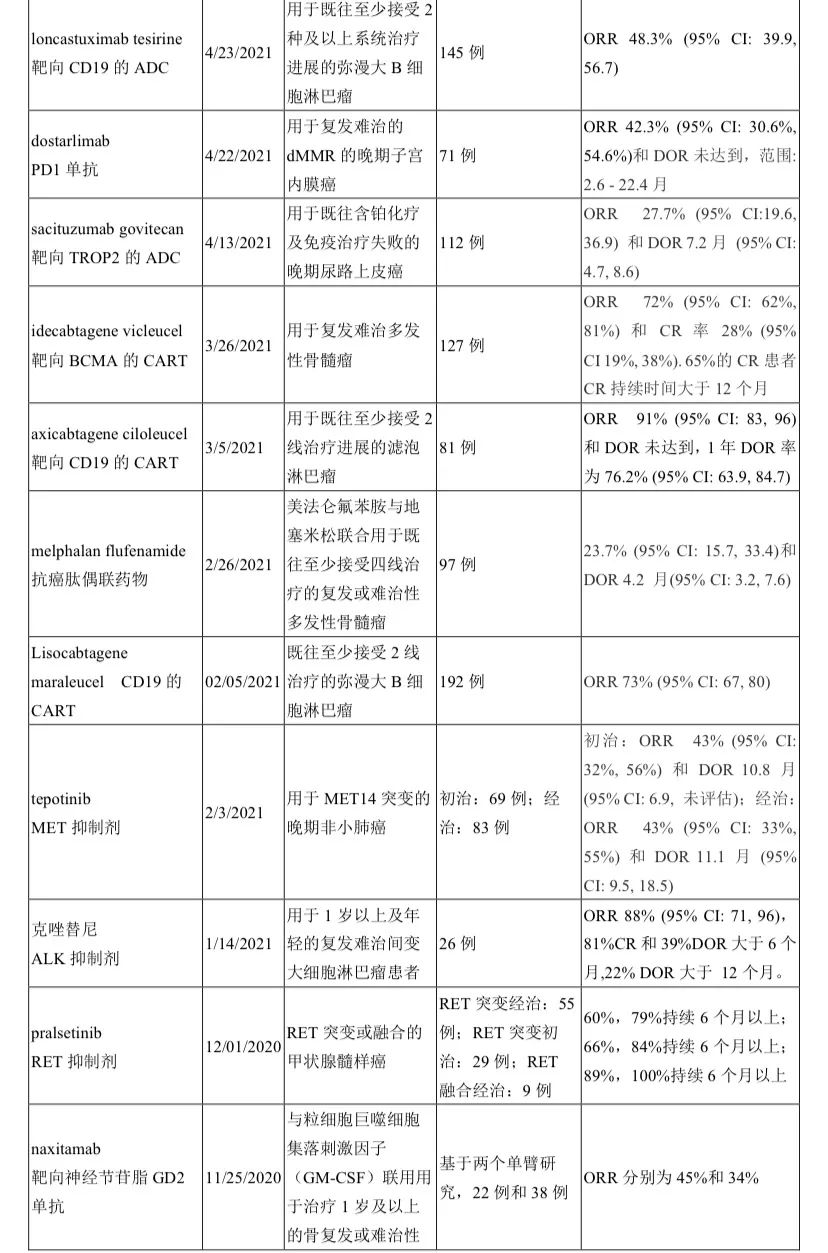
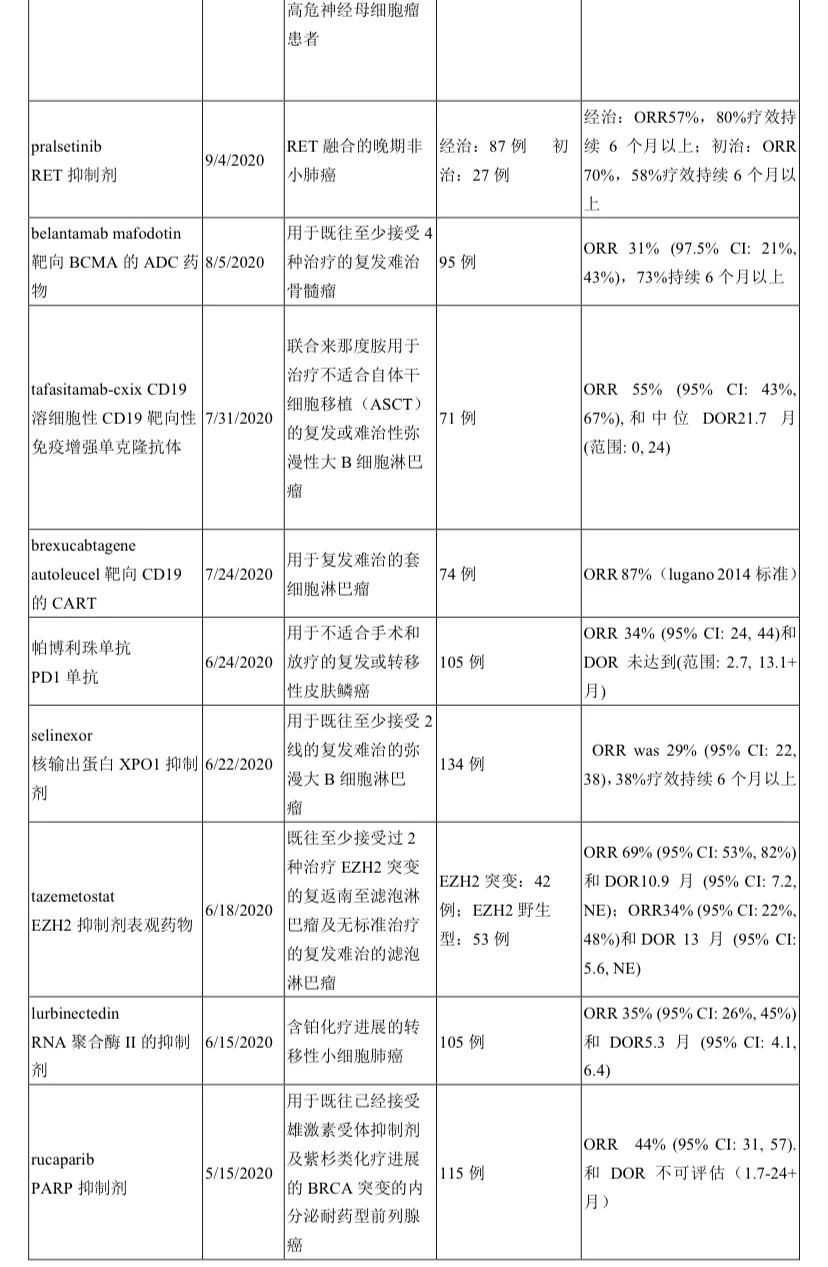
小编整理了FDA官网上 2020年至2021年截至10月17日公布的114份FDA获批通知(其中3份是非治疗),逐一翻阅,共统计出41种新药/新治疗产品时通过单臂研究获批上市,占比率高达36%!汇总见下表。
药物类型涵盖靶向、PD/PDL1免疫、CAR-T、ADC、双抗、表观药物、以及化疗,尽显了目前全球新型抗癌治疗的新方向。
之所以说治疗,是指目前不仅在治疗机制上不断创新(如靶向之外出现免疫(检查点免疫、过继细胞免疫)、表观),更是在药物构型上创新(靶靶结合的双抗、化靶结合的ADC等)。
除了国内熟知的这些靶点外,出现了更多新颖靶点如TF、HIF-2α、GD2、XPO1、肽藕联物(氨肽酶)等。FDA的这份单臂清单也是我们国内药企产品选择的参照标杆。
实体瘤中,除了常见癌肿(肺癌8种;尿路上皮癌2种;宫颈癌、子宫内膜癌、前列腺癌、甲状腺髓样癌、尿路上皮癌、胆管癌各1种)外,“非常见”的癌肿也占据重要地位,比如晚期系统性肥大细胞增多症、神经内分泌肿瘤、神经母细胞瘤、卡珀西肉瘤、皮肤鳞癌、上皮性肉瘤、纤维神经瘤等,可谓罕见病中另辟蹊径。
需要提一句的是,单臂上市的产品不只是首次上市的新药,还包含部分老药新癌肿的扩展、老药更优剂型的推广。
除了特征梳理外,小编也借助FDA获批的这份单臂上市产品清单,来梳理一下单臂上市申请注册的关键点,文末附带了单臂上市的CDE文件与重要文献解读,读者朋友们可以政策结合数据来看,加深理解。
①单臂解决的人群主要是复发难治的疾病患者,无论是无药可用的罕见疾病还是常见疾病的后线,在国内外的上市药物中均可见。因此,一种新药的快速上市,适应症的选择非常重要。但是CDE也指出对复发难治的人群选择一定要科学,不可模糊错判,可以细读一下。
②研究的主评疗效:有效性是主要终点(实体瘤的ORR,血液肿瘤可能特异,看CR),而随着一些新型疗法的出现(如免疫的长效性显著),疗效持续时间DOR也成为了主要主评终点。
因此,读者朋友们可以看到上表中近两年的新药几乎均已ORR和DOR未上市注册研究的主评终点。
③何为更优!单臂上市并不是没有对照,它的对照是采用历史数据的隐形对照,因此在初期考量药物单臂上市及样本量计算时,要搜寻好清晰可靠的历史疗效数据,在这里,CDE指出这个历史对照必须是当下、同一背景环境下的数据,这一点,在新药突飞猛进、疗效时时更新的今天,增加了不少挑战。
有了参照,优多少算是合格?指南指出,主要疗效终点的 95%置信区间下限高于约定的历史对照疗效是支持上市申报的必要条件。在样本量不大的单臂研究中,数据绝对优效和一致稳健性很重要。
④样本量:在CDE指南中,对于样本量未作出具体数据规定,提及对常见的疾病可按50-60例,罕见的可减少至20-30例。上表中例数差异显著,十几、二十几的也并不少见。
⑤安全性,对于上述疗效的注册研究,样本量要求不高,但对于需要证明药物的安全性来说,300例使用这类药物的样本量要求还是很必要的,因此单臂注册研究,往往需要搜罗使用这类药物的所有健康人和患者,进行安全性分析,以支撑新药的安全性评估。
⑥独立评审员会 (IRC ),保证疗效评估的科学性,可以理解。
⑦确证性后续研究:单臂上市往往是附条件,后续都需进行确证性研究,而且OS依然是证明获益的金标准,因此单臂上市也有可能被撤销,近几年,有越来越多的新药因无法证明后期的获益而撤市。当然并不是OS无统计学获益就一定会被撤,仍然需要积极沟通。
CDE:《单臂试验支持上市的抗肿瘤药进入关键试验前临床方面沟通交流技术指导原则》
会议将基于产品自身的前期临床试验数据,关注产品是否存在严重影响临床研发的安全性问题。
申请人需根据本品的前期临床试验数据,综合临床药理学试验结果,进行进行药效学指标、安全性、耐受性和有效性等多维度分析,提供关键试验拟采用剂量的合理性证据。
随机、盲法、平行对照试验是确证药物安全有效性的金标准,因此,如期望采用单臂试验支持加速批准上市应充分评估其可行性。应充分阐述目标适应症当前的治疗现状、历史数据的可靠性、亟待解决的临床需求,以及本品已获得的安全有效性数据,评估关键试验拟采用剂量的有效性对临床获益的预期,综合考虑。
原则上,单臂试验适用于在严重危及生命且缺乏标准治疗手段的难治疾病背景下,疗效突出的单药治疗;将重点评估疗效是否具备显著优于现有治疗的潜力,值得采用单臂试验加速研发。对于疗效突出的抗肿瘤药,申请人可自评估符合“突破性治疗药物”的条件后,参照《突破性治疗药物审评工作程序》申请认定。
由于有效性是考虑是否可以采用单臂设计的关键要素之一,因此,重点关注本品前期有效性数据是否足以支持进入关键试验;不同适应症各具特点,因此对前期有效性数据样本量的要求有所不同;建议申请人在沟通交流时,提供目标适应症发病率情况,在发病率相对较高,患者数量相对较多的人群,例如晚期非小细胞肺癌中的 T790M 突变患者,通常要求具备 50~60 例关键试验拟采用剂量受试者的有效性证据,如果属于发病率较低的疾病类型,例如淋巴瘤中一个罕见的特定亚型,也应要求具备 20~30 例受试者的有效性证据。
此外,进入关键研究前所需有效性数据的样本量,还将结合适应症目前治疗现状综合考量。将根据产品前期的数据评估其疗效是否突出,进而评价是否可以支持开展关键单臂试验。经讨论符合进入关键单臂试验条件的抗肿瘤药,将进一步讨论如下内容。
明确关键单臂试验中对于“复发/难治”受试者的定义。定义的要素通常包括:既往接受过的治疗方案、治疗线数、治疗周期数(某些恶性肿瘤还需明确既往某药物累积剂量)、疾病复发/进展时间或距离末次治疗时间(某些恶性肿瘤需注意区别早期复发和远期复发人群)。
关键单臂试验的人群定义,应体现出“充分治疗、缺乏标准治疗手段”的特点。例如,对于复发性经典型霍奇金淋巴瘤,目前要求在复发前 12 个月内至少接受过二线化疗;距离末次治疗 12 个月以上复发的人群,仍有可能通过重新接受前一线治疗获得缓解,因此不应纳入关键单臂试验。再如,对于难治性经典型霍奇金淋巴瘤,目前要求疗程≥2 周期未达到部分缓解(partial response,PR),或者疗程≥4 周期未达完全缓解(completeresponse,CR),如受试者在某化疗方案经过1 个疗程后因疗效不佳或毒性不耐受之外的原因更换方案,
该治疗方案不应视作一个治疗线也不应被判定为“难治”。
如依据明确的疗效预测生物标志物选择人群,例如计划入组带有特定疗效预测生物标志物的跨瘤种受试者时,需提供目标疗效预测生物标志物的概念验证试验结果 。
-
终点选择:单臂试验通常选择客观缓解率(objective response rate,ORR)作为主要终点。在实体瘤中,ORR 一般由部分缓解(PR)及完全缓解(CR)构成。部分肿瘤(如血液肿瘤)可能有所不同,因此需在沟通交流中明确主要终点的具体定义。
在考虑终点指标的选择时,还应充分考虑不同类型药物的作用特点,例如免疫治疗药物在目前尚未明确生物标记物细分患者的情况下,难以获得与分子靶向药相似的高缓解率,因此,仅考虑 ORR 和持续时间并不能充分反应有效性,还应关注药物持续作用带来的获益和风险,将 12 个月或是更长时间的生存率、持续缓解时间(duration of response,DOR)、无进展生存时间(progress free survival,PFS)等作为重要的有效性指标予以评估。
评估标准和访视计划:根据目标适应症和药物特征,选择国际上广泛采用的疗效评估标准,并在方案中予以明确。根据适应症的临床实践和产品特点制定访视和随访计划。
-
疗效的历史对照:根据目标适应症历史对照数据的依据,明确关键单臂试验有效性的统计假设,并与中心达成共识。
-
有效性分析集:单臂试验的有效性分析集应以 ITT 原则为参考;沟通交流中需明确有效性分析集的定义并达成共识。
-
敏感性分析要求:结合适应症和产品特征,必要时将对敏感性分析提出要求。申请人最终应对有效性结果,开展包含但不限于沟通交流中明确的敏感性分析。
对于采用单臂设计作为关键试验支持上市的情况,需充分考虑对安全性评价的要求。在采用单臂设计时,如按照统计学假设计算出的样本量较小,需考虑为满足安全性评价增加样本量或者启动其他研究。
通常单臂试验要求以IRC评估的有效性作为主要疗效终点。申请人需提供 IRC 的评估标准、评估机制和评估流程,参照中心已发布的《抗肿瘤药临床试验影像终点程序标准技术指导原则》合理设计,保障评估结果的独立性、客观性和稳定性。会议将讨论 IRC 章程的科学性。
在关键性试验开展前,应对未来药品上市许可申报的条件予以明确,通常关注以下几点:
主要疗效终点的 95%置信区间下限高于约定的历史对照疗效是支持上市申报的必要条件;将根据疾病特点和不同产品机制的特征,对有效性评价的随访时间提出要求,并以此明确递交药品上市许可申请时应具备的最低随访时间,以及审评过程中需要更新递交的数据。
根据不同适应症的发病率情况,明确递交药品上市许可申请时对安全性暴露量的要求。通常情况,在拟上市及以上剂量的暴露超过 300 例患者,方能观察到药物在该适应症的常见不良反应及总体安全性特征,用于制定说明书中安全性相关内容及上市后风险控制计划;暴露人群的组成(如暴露人群是否为同一适应症人群,是否为同一种族人群等)以及暴露人群接受的剂量(如是否要求为 RP2D 及以上剂量等),将根据不同适应症的特征具体要求。
例如,治疗晚期非小细胞肺癌的药物在递交上市申请时目标剂量及以上的暴露量需具备 300 例;在一些发病率相对较低的瘤种和人群,不对暴露人群的组成和剂量作出具体要求,但通常整体暴露量仍需满足 300 例。
将讨论拟支持完全批准的确证性试验的方案设计。在本次沟通时,至少应具备初步的试验设计及实施计划。计划同步开展确证性试验的申请人,建议在本次会议时准备详细的确证性试验的方案及实施计划。
确证性试验一般采用随机对照研究设计;对于不适合开展随机对照研究作为确证性试验的情况(例如采用篮子试验设计支持跨瘤种适应症申请时),可考虑开展多个单臂试验作为确证性试验。
结合产品特征,对其他可能影响研发和上市的潜在问题(如是否需要伴随诊断等),予以讨论。
《中华肿瘤学杂志》2018 年 1月第40 卷第 1期:单臂试验支持抗肿瘤新药注册的考虑
虽然 SAT存在上述局限,但当满足了充分的历史数据、难治的疾病背景、较高的客观缓解等以下条件时,即便依靠单臂数据仍可判断获益来自于治时,以SAT支持新药注册是可行的。
SAT的历史对照需来自高级别循证医学证据,应具备相近年代 、相似疾病背景和足够样本量的特点。推荐来自单个RCT 、系统回顾或meta 分析。例如我国肝细胞癌病因主要为乙肝病毒感染,欧美为丙肝病毒感染。这导致我国患者预后差。
SHARP研究为索拉非尼在欧美肝癌患者中开展的随机、安慰剂对照的Ⅲ期临床试验,一线Child—pugh A级的晚期肝细胞癌患者随机接受 1:1索拉非尼(400 mg,口服,2次/d )或安慰剂治疗,主要终点为OS ,索拉非尼和安慰剂的OS分别为10.7和7.9 个月,风险比HR为0.69 ,P <0.001。Oriental研究为索拉非尼亚太桥接试验。设计和入组和排除标准同SHA RP研究。Oriental研究中索拉非尼组和安慰剂组的OS分别为6.5 和4.2个月,H R = 0.68,P = 0.014。因疾病背景差异,Oriental研究索拉非尼组的OS甚至低于 SHARP 研究的安慰剂组,但由于为 RCT 设计,两者风险比一致,如选择SAT,Oriental研究或难以判断索拉非尼疗效。
SAT应在复发难治、医疗需求亟待满足的疾病背景下开展。SAT受试者应为目前标准治疗失败、已无标准治疗或标准治疗效果不满意的病患人群。有标准治疗时,应在充分考虑该治疗的可获及性后,可考虑单臂策略。
由于无对照设计以及历史对照的不确定性,SAT 需慎重考虑主要研究终点。ORR定义为肿瘤缩小达到一定比例并维持一段时间的患者的比例,包括疾病稳定患者的比例复发难治的肿瘤患者在常规治疗下极难观察到客观缓解,可用疗效解释肿瘤的客观缓解。而PFS包括了疾病稳定 ,单臂设计下不能排除肿瘤自然病程的影响;OS还受后续治疗的影响。
因此,选择PFS等时间终点或难以判断治疗获益,SAT常采用ORR为主要终点。
确定终点后,应预设目标值目标ORR应在系统回顾疾病背景、同类产品和本品的临床数据后制订 ,并与监管机构沟通由于SAT是一种有条件的加速批准策略,意味着结果具有不稳健性。监管机构将承担 I 类错误风险( 即批准一个实际无效的品)。因此,仅当ORR显著提高时,即便为SAT也可判断获益来自于治疗。
较高的ORR指与历史对照相比,有较大提高,不要求ORR成倍增加 ,但95%CI下限应高于历史数据上限。
应兼顾缓解时间,较长的缓解时间是SAT获批的重要因素。Atezo在二线转移性膀胱癌患者中的ORR虽仅为 14.8%,但80%患者的缓解持续时间超过6个月,13%患者的缓解持续时间超过12个月,基于Atezo 持续的ORR。FDA批准了其转移性膀胱癌二线治疗的适应证。在现阶段OS仍为肿瘤患者临床获益的金标准。
ORR为单臂策略下 OS 的替代终点,持续的缓解时间将增加治疗获益的稳健性,同时兼顾安全性和耐受性信息。
当研究以影像学数据为终点结时,推荐选择独立影像评审委员会 (IRaC ) ,单臂设计应选择IRaC评估的ORR为主要终点;当研究者评估的ORR为主要终点时,以IRaC 评估终点进行敏感性分析。设置 IRC或IRaC的目的是为了减少研究者偏倚。
由于样本量少.SAT的安全性数据有限,不能充分暴露药物的安全性信息,不利于对药物安全性风险的控制。因此,申办方应考虑研究计划,在注册时提供足够的安全性信息。
富集试验设计提高了研发效率。监管机构鼓励开展以生物标志物富集人群的新药研发,但企业需完善伴随诊断方法,例如,克唑替尼同期批准的ALK融合突变检测方法(荧光原位杂交和Ventana方法) 。
回顾SAT的成功案例,或可发现部分获批药物的关键研究来自2项SAT。如克唑替尼和奥西替尼,2项SAT结果相似并相互验证,提高了稳健性。
1. 2020年至2021年FDA批准单臂研究上市药品数据;
2. CDE:《单臂试验支持上市的抗肿瘤药进入关键试验前临床方面沟通交流技术指导原则》;
3.2021 CSCO杨志敏部长《抗肿瘤新药附条件批准的审评考量》;
4. 《中华肿瘤学杂志》2018 年 1月第40 卷第 1期:单臂试验支持抗肿瘤新药注册的考虑。
版权声明/免责声明
本文为授权转载文章,版权归拥有者。
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系(电话:13651980212。微信:27674131)。衷心感谢!

11月11日,一个非常特别的日子!
我们在上海张江,一个非常特别的地方,
探讨一个非常特别的话题!
我们不见不散!

 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
















 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 





