来源:药渡,作者:东华帝君
原文发布时间:2018-02-20
美国食品药品监督管理局(FDA)药审中心(CDER),之所以成为了全球最具权威的药审机构,与其整个系统的科学性、职能部门的高效率、工作人员的严谨性密不可分。而整个药审中心的合理建设,得益于建立在一件件法案的顶层覆盖之下,才能使之健康、快速、可持续的运营下去。在众多法案之中,1992年开始的《处方药使用者付费法案》(The Prescription Drug User Fee Act ~ PDUFA),被业界称之为“美国现代新药审评的基石”,开创了新药审评审批的新纪元,且直至今日,仍在更新使用当中,值得业界学习。
1992年之前,由于FDA缓慢的药品审批状态,使药品上市晚于其他国家和地区,美国患者对此极为不满,认为FDA将美国消费者置于不利地位,这一现象,被制药行业称为“药品上市迟滞”。
过长的审评时间,不仅耽误患者使用这些药品治疗,还影响药品生产商收回研发成本。FDA在当时估计,审评完成每迟滞1个月,带给生产商的平均损失达1千万美元。但在1992年,FDA药品审评人员配置严重不足,这就自然客观的存在审评流程不可预测、审评速度慢等弊端。
当时的美国药品生产商协会(PMA)主席Gerald J. Mossinghoff(曾担任美国专利局局长,该机构完全由专利申请付费提供资助),在FDA是否可以收取企业付费问题上率先打破僵局。认识到FDA经费不足将对产业界造成损害,试图在制药行业与FDA之间找到对双方均有益处的共同点,并提出4项具体条件。但出于总统竞选的压力,布什直到大选前1天才签署该法案,同意通过收费筹集的资金将用于补充而非替代国会下拨给FDA的拨款,PDUFA得以通过。
PDUFA授权FDA收取3种费用:新药申请费(1992年~10万美金)、设施费(1992年~6万美金)、产品费(1992年~6千美金)。这些费用逐年上调,在PDUFA第VI版新药申请费已经上升为242.1万美元了,后面这两种收费由于是基于相对稳定的基数(不管是药厂数目还是上市的药品数,都不会有太多太大的突然变化),保证了FDA有相对稳定的经费来源。
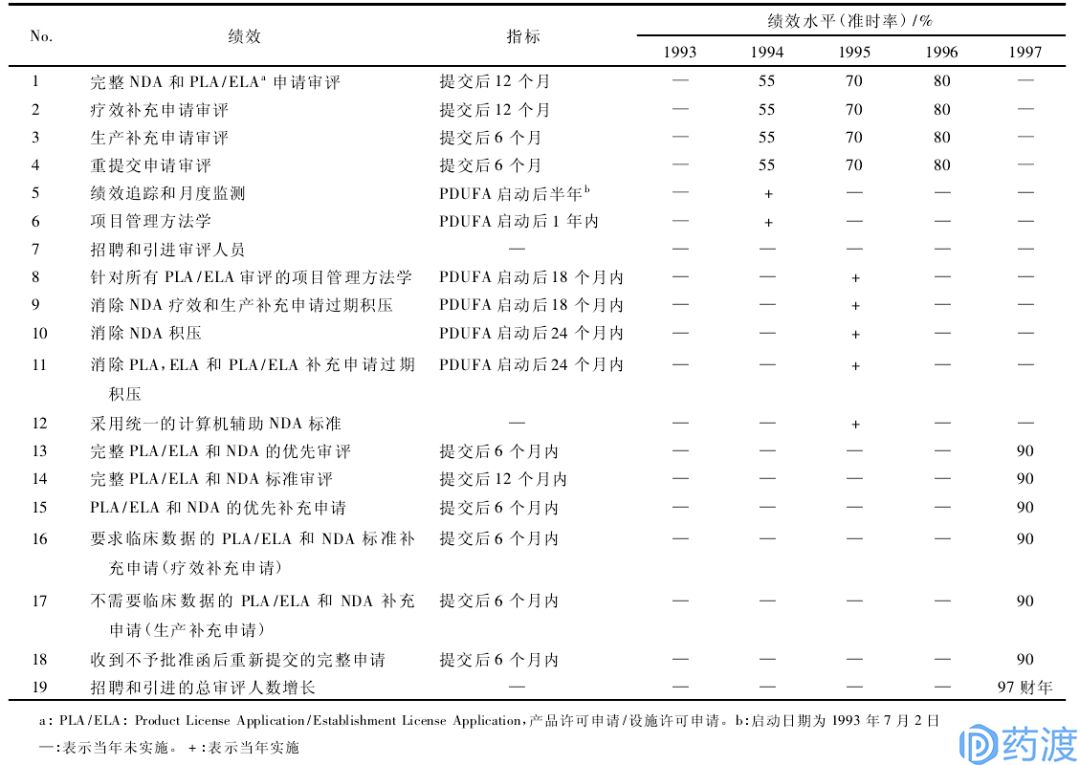
1992~1997的5年间,PDUFA-I取得了极大的成功,FDA在没有降低新药审评标准和增加国家财政拨款的前提下,处理了堆积如山药品申请,探索了新的审批机制和过程,设立了药品审查过程中的各个工作时限,圆满完成了PDUFA-I所设定的各个目标,平均审批时间从实施前的21个月/件,下降到1997财年末的12个月/件。PDUFA-I的顺利实施也增加了美国国会及FDA的信心,至今已更新至2017年的第VI版。

在1992年PDUFA法案中,FDA同意改善药品审评的具体目标,制定了优先审评和标准审评两个层次的审评时间(分别为6个月和10个月)。各项审评绩效指标采取逐步细化、循序渐进的方式,不要求一步到位。规定允许收取费用用于“人用药品申请审评必需的活动”,包括人用药品申请和补充申请、批准人用药品申请或陈述申请中的具体缺陷的决定书、作为待批人用药品申请或补充申请审评的处方药设施检查、公共健康服务法第351节规定的生物制品放行设施取证申请审评,以及监测与人用药品申请审评相关的研究。伴随着法案的实施,新药申请量得到大幅度提升、被拒申请得以减少、批准率提高、批准时间更快,以及审评人员成倍增加。
表2:PDUFA-I的绩效目标(1993~1997财年)
在PDUFA-I第一个5年的显著成果基础之上,第二个5年计划中,PDUFA-II重点放在加快药品研发和申请审评流程,规定了更为细化和严格的绩效目标,药品审评流程更加透明,以及和制药公司、患者、权益团体的更多沟通。与此同时,PDUFA-II还首次要求FDA成立科学咨询委员会,负责在研究、临床试验、上市批准方面为FDA提供专家意见和建议。在此期间,FDA在全球首个批准的药品占40%,意味着药品上市迟滞问题已初步得以缓解,美国消费者可更快更早地获得药品。
表3:PDUFA-II的绩效目标(1998~2002财年)
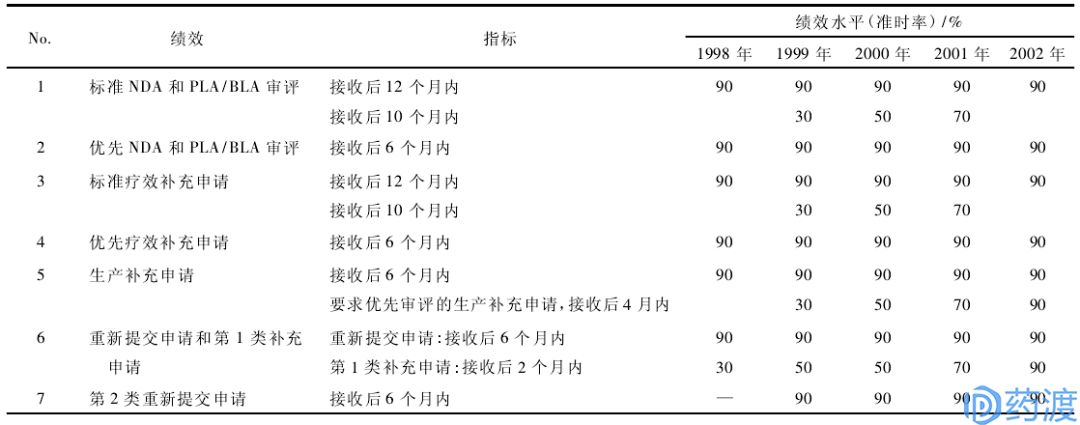
重点:细化审评流程,扩充从PDUFA中收取的资金的运用范围,首次将范围扩展至临床前研究和批准后上市的头3年。
允许FDA使用PDUFA提供的资金用于收集、开发和评价批准后3年的上市后安全性资料,这一变化使得FDA可以将负责监测上市后药品副作用的人员增加1倍,并使FDA可以开发记录药品使用的数据库。除此之外,PDUFA可根据工作量变化调整预算目标、与利益相关方召开相关会议、收集开发审查一定范围内的安全性数据、允许选择独立的咨询机构参与FDA对研究活动的审评、批准两项旨在强调没有现成治疗药品可用的严重和致命症状的快速通道药物申请的滚动性审评试验计划、鼓励公司在NDA/BLA前会议中纳入风险管理计划、允许筹集资金用于“围批准期”(即批准后2~3年)的风险管理监督、制定“首轮”初步审评等等。
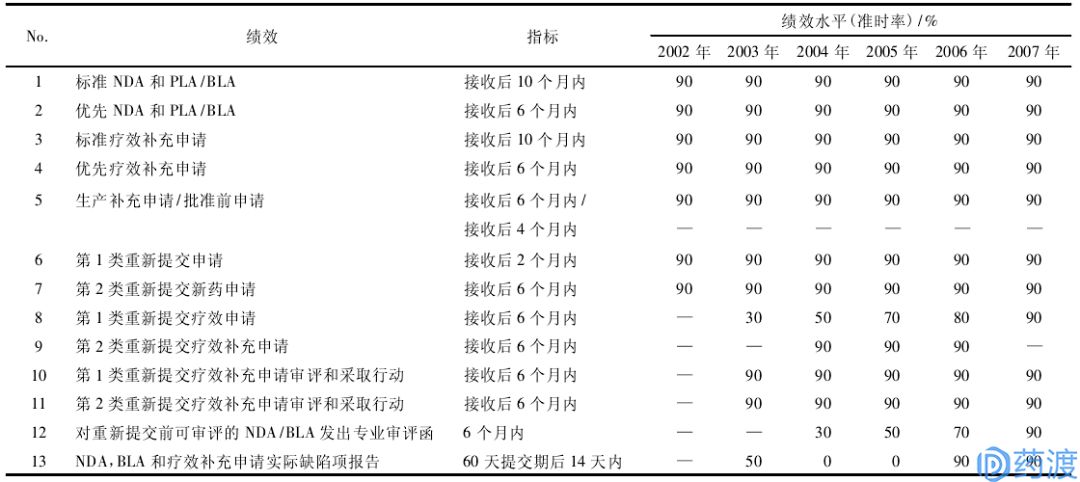
表4:PDUFA-III的审评绩效目标(2003~2007财年)
重点:PDUFA-IV突出了对安全性的要求,有了一些技术性变化,并引入了新的要素。
有了PDUFA-III的成效,PDUFA-IV在拨款水平的基础上,增加了反向调节点;增加用于药品安全性的付费(总额达到2. 25亿美元);取消对采取批准后措施的年份和时间限制(更大范围的使用募集);取消了对上市后活动的3年限制,将上市后安全范围扩展至PDUFA所能支持的范围;批准对处方药电视广告有关的咨询审查开展评价和收取有关费用;《联邦食品、药品与化妆品法中》写入一些与PDUFA有关的关键内容;加强HHS部长与公众间在FDA和产业界谈判问题上的沟通等等。
表5:PDUFA-IV的审评绩效目标(2008~2012财年)

重点:NDA+原创性BLA创立新计划、透明度、沟通。
在PDUFA-IV的基础上,PDUFA-V对新分子实体NDA和原创性BLA绩效目标方面创立了新的计划,强调审评过程更高的透明度和与申请人之间的沟通(如原始申请提交、74天函件、中期交流、专业审评函、期末会议、检查等)。建立风险-收益,以指导药品审批,将REMS标准化,力图使FDA和产业界逆转近年来药品研发和批准方面出现下降的趋势,最终使美国公众获益。
表6:PDUFA-V的审评绩效目标(2013~2017财年)
重点:药物研发以患者为中心、继续推进罕见病与突破性疗法的审查、增加对药物安全性方面的投入、探索真实世界的证据用于决策、确保FDA能够聘请保留优秀的科学工作者。
在刚刚过去的2017年8月18日,美国总统签署了《食品药品管理局重新授权法》(FDARA)。PDUFA随之被重新授权,PDUFA-VI正式诞生。此前,2015年9月~2016年2月期间,FDA已经同制药业代表、患者、医生及其相关者进行磋商,制定了PDUFA-VI的强化措施,于2016年12月正式向国会递交提案。新法案的授权,确保了2018~2022将继续获得稳定持续的资金支持,从而帮助FDA更好的履行保护和促进公众健康的使命,帮助患者推出更好的新药。本次PDUFA-VI是建立在先前法案的成功经验基础之上,一些重点领域将被更多关注,包括:药物开发要更多的以患者为中心、继续推进罕见病与突破性疗法的审查、增加对药物安全性方面的投入、探索真实世界的证据用于决策、确保FDA能够聘请留住优秀的医药工作人员。

PAUFA-I覆盖范围主要集中在NDA至上市批准,这一阶段主要解决了审评资金、申请积累的问题;PAUFA-II覆盖范围扩展到了IND阶段,进一步缩短了审批时间、制定了各种目标,增加了储备资金;PAUFA-III扩展至临床前研发阶段和上市后3年,实现了GRMP互动、上市后检测,进一步增加了资金;PAUFA-IV进一步扩展了上市后监测的时间范围,增加与稳定资金、增强上市前审评、革新上市后监测;PAUFA-V范围同IV,但在细节上进行了加强,如增加了资金、强化上市后监测、提高透明度与可预见性;PAUFA-VI扩展到研发思路方面上,强调以患者为中心、探索真实世界、同时确保审评人员的长期聘用。至今,PDUFA可以说,完美的逆转了药品上市迟滞、显著缩短了新药审评时间、弥补了国会拨款不足、增加了审评人员数量、加快了获得性治疗药物的速度、制定了行业指南、权衡了获益与风险,值得国内深入学习。
PDUFA法案,自签署至今,已整整经历了5个“5年计划”,从民众的抱怨、企业的催促,到PMA的介入、法案的签署,这其中的困难程度可想而知。但面对于此,美国FDA并没有选择推诿,而是敢于大刀阔斧的去改革,去碰触利弊,去解决问题,从而实现了审评的加速、药品的快速上市,使美国民众生命健康受益。这其中,无论是法案,还是解决问题的思路,都值得其他国家和地区针对本国特点进行改良,如此,则不仅提高了本国药审机构的效率,更重要的是使本国人民可以更好的使用到当下最为有效的药物。
参考:
1. What you need to know about PDUFA VI。http://catalyst.phrma.org/what-you-need-to-know-about-pdufa-vi.
2.https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM511438.pdf
3. http://phrma-docs.phrma.org/sites/default/files/pdf/Factsheet-PDUFA-VI.pdf
4. 大招频放的中国食药监局能从这项美国法案中学到什么?知识分子
5. 从PDUFA I到PDUFA V~FDA通过法规体系的完善实现新药审评的持续改进,CNKI
本文为授权转载作品,仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
文中图片为授权正版图片,或来自微信公共图片库,或取自网络

 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权





 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来!