

所有抗肿瘤药物治疗的最终目标是改善以病人为中心的终点。这些“硬性”终点对患者来说具有很好的临床获益,它们包括总生存期(OS)的延长、生活质量(QoL)的提高,或者两者兼而有之。OS既可以反应治疗的疗效又可以体现安全性,且OS终点客观精确可测,并有死亡日期提供依据,在终点评估时不会出现偏倚。所以说,OS被认为是迄今为止肿瘤临床试验中评估药物疗效最可靠和临床意义最大的终点,是临床研究的“金标准”。
然而,临床试验选择OS作为主要研究终点往往需要大样本量和长期随访,因而会增加临床试验的成本,并可能延迟真正有效的治疗在临床上的应用。同时,OS 容易受到后续治疗的影响,因此可能在一定程度上掩盖了初始治疗的真正效应。此外,由于对肿瘤疾病认知的不断进步,尤其是近年来各种新的治疗模式横空出世,传统的化疗之外,靶向治疗和免疫治疗,让晚期肿瘤治疗逐步从一线治疗、二线治疗推进到N线治疗的可能性,治疗的排兵布阵逐渐丰富起来,但是想要做出OS获益的研究却变得越来越难。正是基于上述多个因素的存在,近年来很多临床试验研究中采用了大量OS替代终点(surrogate endpoint),用于新药研发的疗效和安全性评估。
何谓替代终点?
美国国立卫生院(NIH)将其定义为“一种旨在替代临床终点的生物标志物”。FDA认为临床试验的替代终点是“直接测量患者感觉、功能或生存状况的实验室测量或物理指标,作为临床意义终点的替代品,可以预测临床治疗效果”。与临床终点相比,替代终点通常可以更早测量,而且需要更小的样本量和更短的随访时间。在肿瘤药物治疗中,衡量药物抗肿瘤活性的生物标志物,如客观应答率(ORR)和无进展生存期(PFS),均被提出并且作为临床试验的替代终点进行评估。
据1995年至2009年肿瘤随机对照试验的综述显示,在1995年至2004年间,49%的试验以OS为主要终点,但到2005年至2009年,这一比例已降至36%;而应答率也从14%下降到6%;但是,以time-to-event的终点(如PFS)占比远远超过了预期,从26%上升到43%。据另一篇文献显示,自2009年至2014年,FDA共批准83种不同肿瘤适应症的药物上市,其中55(66%)种适应症的获批是基于替代终点(31种基于ORR批准,24种基于PFS批准);按照批准方式来说,采用加速批准的25种适应症100%都是基于替代终点,而采用传统方式批准的30种适应症种51%是基于替代终点。
那么,在FDA以替代终点批准的上市药物中,替代终点的改善究竟能否真正替代OS获益?是否可以表明患者生活质量的提高呢?下面我们分别来看:
2019年1月,在《European Journal of Cancer》公开发表的一项系统评价,回顾性的分析了肿瘤治疗中替代终点与总体生存率之间关联强度。
这篇综述有几个十分明显的优势:
(1)这是截止目前所有已发表相关研究中对于肿瘤替代终点与OS关联性研究中规模最大、最新最全面的分析。同一团队曾经于2015年发表过该相关性文章,此篇截取时间至2018年,更新并增加了近几年通过替代终点获批的试验,其中2015年及以后共发表文章37篇(47%)。
(2)通过Google Scholar和PubMed搜索所有符合条件的随机对照试验meta分析,只纳入基于trial-level (一级)的分析,因为这是替代终点有效性验证的最高证据级别;
(3)对药物治疗的方法没有限制,包括转移、新辅助、辅助或局部进展,免疫治疗相关性作为一个单独的类别被纳入,因为越来越多的人认识到,替代指标和OS之间的关系对该类药物是特殊的;
(4)采用替代终点与OS之间的关联性强度分级方法采用的是德国卫生保健质量和效率研究所( IQWiG)改进的量表体系:低强度相关(r ≤0.70),中强度相关(0.70< r <0.85),高强度相关(r ≥0.85)。
综述最终结果如下:
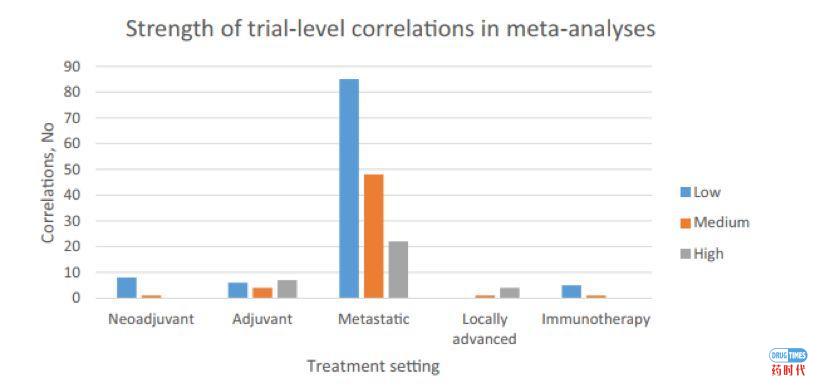
(1)按照不同的适应症和发表日期显示了试验级替代终点的证据演化图(AERO图)。根据替代终点与OS之间的相关性强度,对节点进行颜色编码(绿色—相关性高;黄色—相关性中等;红色—相关性低。节点的大小代表每个meta分析中包含的试验数量)。由AERO图发现,大多数相关性发生在转移性治疗中(n=65)。更为重要的是,在上述89项验证研究中,仅有11项(12%)显示了高相关性,9项(10%)显示了中等相关性,34项(38%)只报告了低相关性,另外还有35项(39%)报告了不同强度的相关性,这取决于使用的替代终点和相关性计算方法。

(2)报告了193项个体试验水平相关性研究,采用了不同的方法计算相关性和不同的替代终点。其中,34项(18%)显示高相关性,55项(28%)显示中度相关性,104项(54%)显示较低相关性。当观察最常见的替代终点时,无论癌症类型如何,PFS个体试验水平分析中,低强度相关性占比为48%(n=40 /83),中强度相关为33% (n=27 /83)和高强度相关性19%(n=16 /83)。相反,在32个使用ORR的个体试验级分析中,29项(91%)为低相关,3项(9%)为中度相关,无高相关。
结论:
在肿瘤替代指标与OS之间的相关性研究中,只有约12%的相关性较强,而88%的相关性强度较低或中等,在这一综合分析中,值得注意的是,有相当比例的癌症药物是通过替代终点获得批准,而在获得突破性疗法认定的癌症药物中,这一比例甚至更高,96%的药物依赖于替代终点。
因此,研究结果表明,肿瘤临床试验中替代终点对于患者OS获益的预测比较微弱,大多数肿瘤学中的替代终点与OS的相关性很低。鉴于替代终点在药物行政审批和临床实践指导中的广泛应用,我们的研究结果表明在基于替代终点得出结论时应谨慎使用。
无进展生存期(PFS)现已成为常用来评估肿瘤新药有效性结局,用于药物的批准上市。然而,PFS的延长是否能够对应肿瘤患者生活质量的提高或者改善呢?二者之间是否具有一定的相关性,这是我们关心的另外一个问题。
2018年12月,在《JAMA Internal Medicine》由加拿大、波兰、日本等国学者共同发表的一项系统评价和Meta分析,回顾性的分析了肿瘤临床试验中替代终点PFS延长和肿瘤患者健康相关生活质量(Health-Related Quality of Life,HRQoL)提高之间的相关性。
数据来源:对癌症患者的随机临床试验进行的系统评价和定量分析,研究者检索了Medline、Embase和Cochrane临床对照试验资料库,检索时间从2000年1月1日~2016年5月4日。试验的入组标准为,研究了口服、静脉、腹腔内或胸腔内化疗或生物治疗,并报告了PFS和HRQoL。
数据采集和分析:研究者使用加权简单回归,检测了治疗组间中位PFS时间(以月计算)差异与治疗组间总体、身体和情绪HRQoL评分(标准化为0~100分的范围,分数越高表明HRQoL越好)差异之间的相关性。
主要结局和测量指标:PFS时间与HRQoL之间的相关性。
结果:
(1)共筛选了35960条记录,最终纳入报告了38项随机临床试验的52篇文献纳入到本分析中(见Figure 1),包括12种癌症类型的13979名患者(乳腺癌7项,大肠癌2项,肺癌6项,黑色素瘤3项,多发性骨髓瘤1项,神经内分泌瘤1项,卵巢癌6项,胰腺癌2项,前列腺癌2项,肾细胞癌1项,胃癌3项,子宫颈癌4项),使用了6种不同的HRQoL工具(EORTC-QLQ-C30 19项,FACT-G 13项,肺癌症状量表3项,EQ-5D 1项,LASA问卷调查1项,Karnofsky评分1项)。
(2)上述RCT临床试验报告的中位随访时间从10.5 ~ 66.0个月,试验干预组的中位PFS为1.8 ~ 33.7个月,报告或测量的HRQoL持续时间为1 ~ 34个月。在38项RCT试验中,其中24项的HRQoL随访时间短于干预组中位PFS。上述RCT试验中3个HRQoL测量完成率是处于13% ~ 100%。
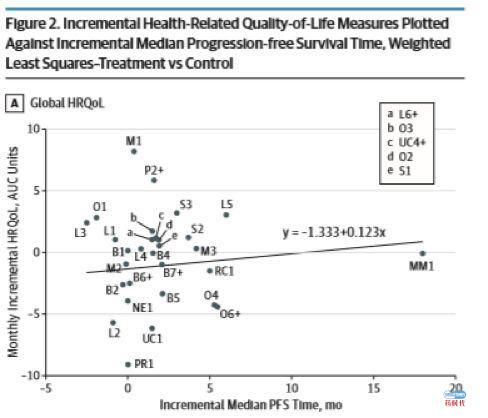
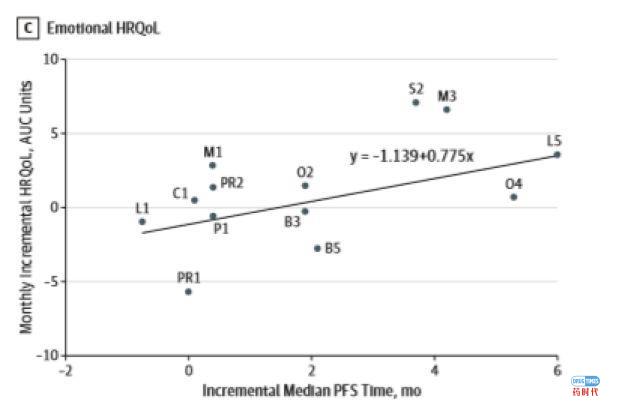
(3)Table 2,Figure 2详细列出了回归分析中使用的研究数据结果。在上述38个试验中,28 (74%)个显示相比对照组,干预组PFS有改善。干预组和对照组间,中位PFS的平均(SD)差异为1.91(3.35)个月。调整了每月值的HRQoL改变的平均(SD)差异分别为总体方面:-0.39(3.59);身体方面:0.26(5.56);情绪方面:1.08(3.49)。中位PFS差异和总体HRQoL改变差异(30项试验)之间的相关性斜率为0.12(95% CI,-0.27~0.52);和身体HRQoL改变差异(20项试验)之间相关性斜率为-0.20(95% CI,-0.62~0.23);与情绪HRQoL改变差异(13项试验)之间相关性斜率为0.78(95% CI,-0.05~1.60)。
结论:研究者未在癌症临床试验中发现PFS与HRQoL之间的显著相关性。这些结果对“延长PFS的干预也会改善癌症患者HRQoL”的假设提出了质疑。因此,为保证患者从癌症治疗中真正获得重要的获益,临床试验研究者应该直接准确测量HRQoL,保证充足的治疗时间和随访期。

上面两部分的回顾性分析的结果告诉我们,肿瘤药物临床试验中替代终点的改善与患者最终OS获益和生活质量的提高之间没有很强的相关性。
FDA有四种特殊的审评路径,其中加速批准都是基于二期临床试验中能够预测临床获益的替代终点的改善,通过加速批准上市的药物按照FDA要求需要在上市后进行三期临床试验疗效验证。那么,我们是否可以通过加速批准上市药物的三期疗效验证性试验来检验上述相关性结论呢?
2018年6月,由FDA血液学和肿瘤学药物审评办公室在《JAMA Oncology》发表了一篇综述,回顾分析了FDA 25年来加速批准的经验,包括了所有的恶性血液学和肿瘤学药物上市后疗效确证试验的结果。
数据来源:从1992年12月11日开始,到2017年5月31日,我们搜索了FDA的数据库,包括了所有经加速批准上市的恶性血液学和肿瘤学适应症的药物。
结果:
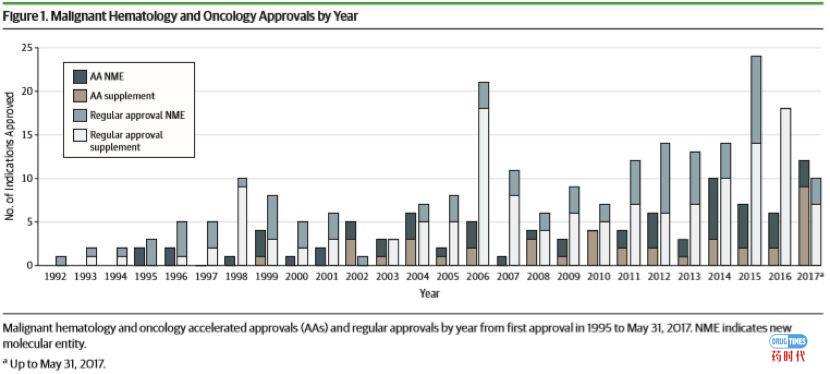
(1)统计区间内FDA共加速批准了64种恶性血液学和肿瘤药物,对应93种新的适应症。在这些加速批准中,有53个是新分子实体。在此期间,174种肿瘤药物适应症获得了常规批准。经批准的恶性血液学和肿瘤学药物适应症按年份和批准途径如图1所示。
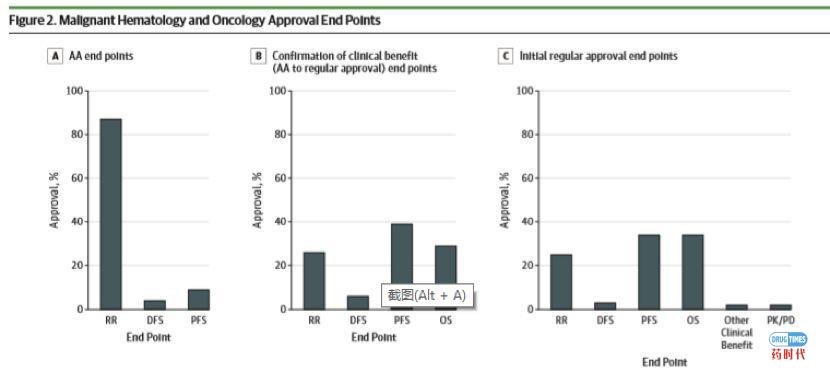
(2)加速批准的试验中,单臂试验占67项(72%),均以ORR为终点指标;而随机对照试验占26项(28%),ORR占比13项,PFS占比7项,DFS占比4项,TTP占比1项, 1项以病理完全缓解作为主要的终点支持加速批准。总的来说,以ORR终点的批准速度最快(81[87%]),其次是PFS或TTP的时间到事件终点(8[9%])和DFS或RFS的时间到事件终点(4[4%])(图2A)。
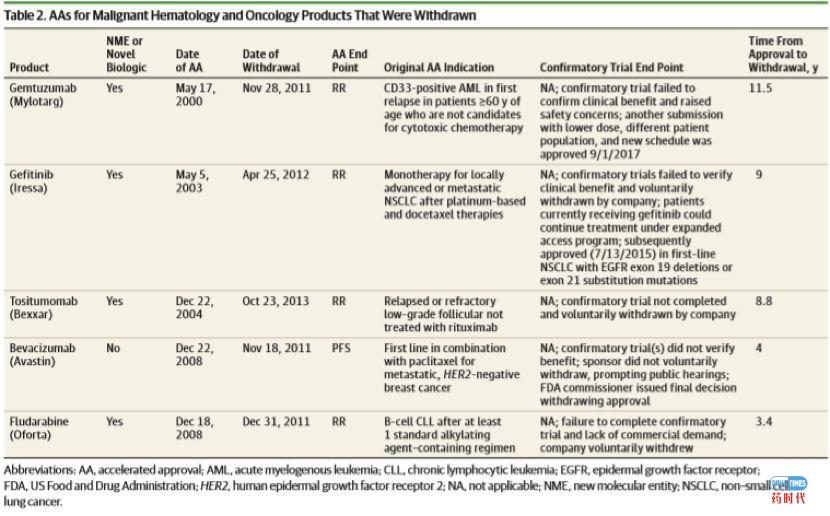
(3)在93项加速批准中,51项(55%)按照上市后的要求进行疗效验证。37项适应症(40%)尚未完成验证性试验。5种加速审批的适应症(5%)已退出市场(表2)。
(4)在51种最初在加速批准上市后并验证临床效益的药物, 主要用于支持定期批准的临床疗效验证的终点是PFS或TTP(20[39%]),其次是OS(15[29%])、支持反应持续时间(包括血液终点)的RR(13[26%])和DFS或RFS(3[6%])(图2b)。
(5)在51种临床疗效验证的适应症中,从加速批准到效益验证的平均时间为3.4年(范围为0.5-12.6年),其中大多数药物在批准时正在进行验证性试验。对于那些在加速批准时正在进行验证性试验的患者,验证受益的平均时间为3.1年。
(6)在37例正在进行验证性试验或尚未验证疗效的适应症中,加速批准至截止日期(2017年5月31日)的中位数时间少于2年(18.5个月),范围从小于1个月到149个月(12.4年)。其中26项(70%)上市时间不足3年,20项(54%)上市时间不足2年。8种尚未证实疗效的适应症已上市5年以上,其中一些适应症针对的是罕见的患者群体。
结论:
作者认为在过去的25年中,经加速批准上市的药物在不断增加,但经上市后疗效验证研究发现,有5%(5种)经加速批准适应症的药物没有得到疗效验证而退出市场,而51项(55%)在上市后完成了疗效验证,另外有37种(40%)适应症尚未完成验证性试验,或是因为上市后时间小于从加速批准到效益验证的平均时间,目前还处于继续跟踪阶段。所以,从FDA监管机构的角度出发,加速批准审查25年的经验表明,该途径是成功的,它平衡了样本量较小和早期临床试验终点相关的不确定性,为那些患有严重或危及生命的疾病的患者提供了更快获得有希望的药物的途径。
但是,若从患者角度出发,抗肿瘤药物治疗最基本和最简单的诉求就是能够活得长,那么我们单独来看FDA 25年来经过加速审批上市的93种适应症药物对于患者治疗OS的影响,又会是什么样呢?
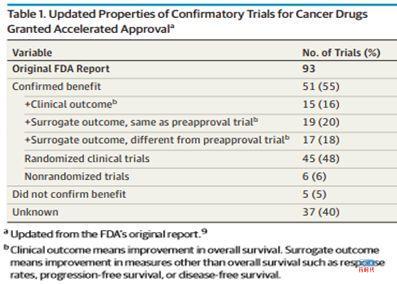
在上述51项已经完成上市后药效验证的临床试验中(Table 1),其中只有15项(16%)在OS获益方面得到了验证,而有19项(20%)采用了与加速批准上市时同样的替代终点指标,又有17项(18%)采用了与加速批准上市时不同的替代终点指标,这些替代终点指标验证都不能直接说明患者在OS方面的获益。
这个结果也可以完全支持前面第一部分中得出的结论,在大多数抗肿瘤治疗药物中,替代终点指标的获益与患者最终OS的获益之间没有很强的相关性。
我们通过一个例子来分析说明,奥希替尼。
加速批准——AURA2研究
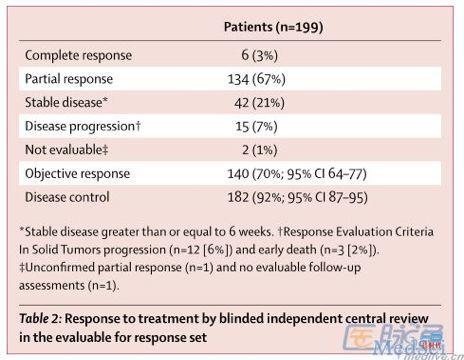
FDA的加速批准依据是基于单臂多中心的Ⅱ期临床试验AURA2研究(ClinicalTrials.gov/NCT02151981),入选患者均接受过EGFR-TKI并出现进展,且T790M抗性突变阳性。患者接受每天一次奥希替尼(80mg),持续到疾病进展,主要终点为总有效率(ORR)。
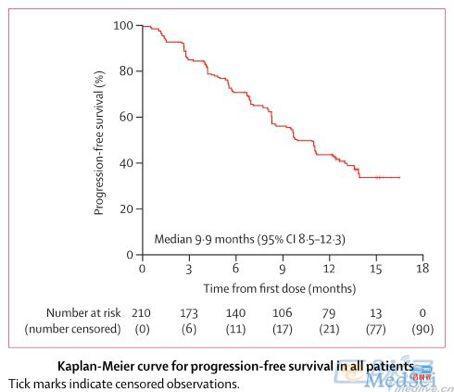
主要结果:ORR为70%(95%CI,64-77),由6例完全缓解(3%)和134例(67%)部分缓解组成。中位无进展生存(PFS)为9.9个月(95%CI,8.5-12.3)。见下图。
三期验证性试验——AURA 3研究
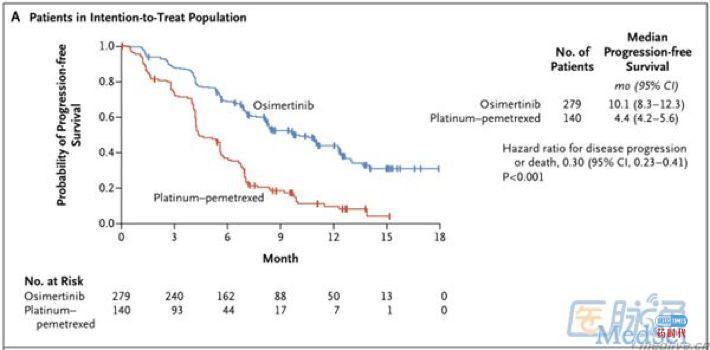
奥希替尼加速批准上市后按照要求进行了三期关键性试验AURA3研究(ClinicalTrials.gov,NCT02151981),将奥希替尼与含铂双药化疗进行对比,评估二者对经EGFR TKI治疗后病情进展的EGFR T790M阳性、局部晚期或转移性NSCLC患者的疗效及安全性。研究者纳入了419例T790M阳性的晚期NSCLC一线EGFR-TKI治疗后疾病发生进展患者,以2:1的比例接受一次奥希替尼(80mg)或培美曲塞(500mg/m2)联合卡铂(AUC5)或顺铂(75mg/m2)。主要终点事件为无进展生存期(PFS)。
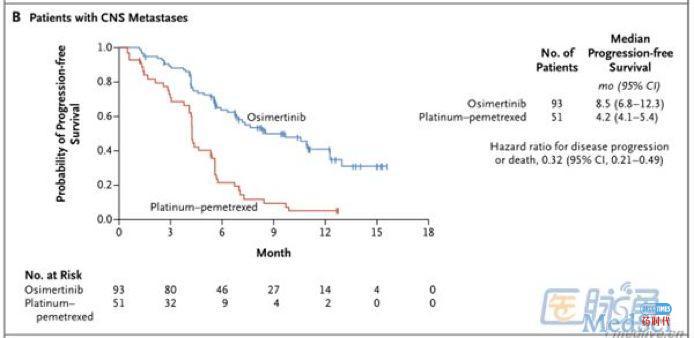
主要结果:mPFS为10.1个月vs.4.4个月(HR 0.30,95%CI,0.23-0.41;P<0.001);有效率(ORR)为71% vs 31%(HR 5.39,95%CI,3.47-8.48,P<0.001)。且在144例病灶转移到中枢神经系统患者中,奥希替尼相对化疗组的mPFS较长(8.5个月vs.4.2个月,HR 0.32,95%CI,0.21-0.49)。而且不良事件也比例较低(23%vs47%)。见下图。
一线治疗——FLAURA研究
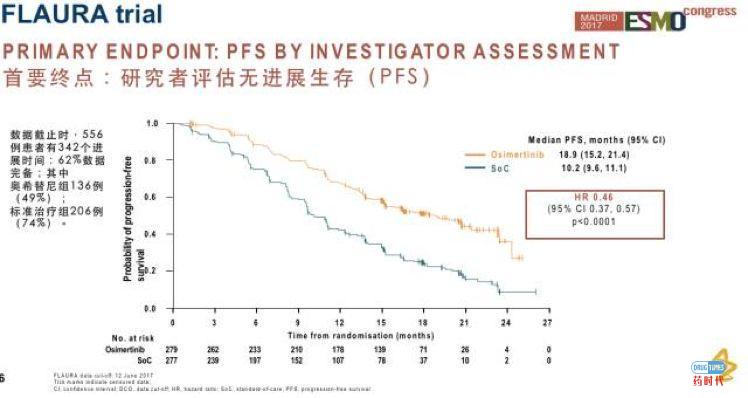
奥希替尼用于T790M阳性的NSCLC患者的一线治疗批准是基于三期临床试验FLAURA研究,共纳入来自30个国家的556名既往未接受过任何治疗的局部晚期或转移性EGFR突变阳性的NSCLC患者,用于评估奥希替尼80mg 每日一次与标准治疗EGFR-TKI治疗(厄洛替尼150mg 口服每日一次或吉非替尼250mg口服每日一次)的有效性和安全性。本研究的主要研究终点是PFS,次要研究终点包括OS,ORR,DoR,DCR,安全性和生活质量评分(HRQoL)。
主要结果:mPFS为18.9个月vs.10.2个月;有效率(ORR)为80% vs 76%;mDOR为17.2个月vs.8.5个月,而且不良事件也比例较低(34%vs45%)。
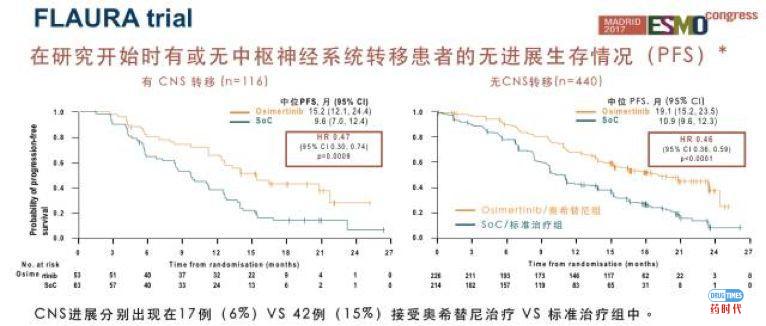
不仅如此,有脑转移的患者,接受奥希替尼治疗,患者无进展生存的获益也优于标准治疗(15.2个月VS 9.6个月),这也再次证实了奥希替尼穿透血脑屏障的良好效果。对于入组时未出现脑转移的患者,奥希替尼组患者的无进展生存期也有明显改善(19.1个月VS 10.9个月)。而且初步统计也发现,奥希替尼治疗组患者出现中枢神经系统进展(包括原发疾病进展和新发病灶转移)的风险也更低。
看到这里,奥希替尼以上临床试验中主要终点都是基于替代终点ORR或PFS。
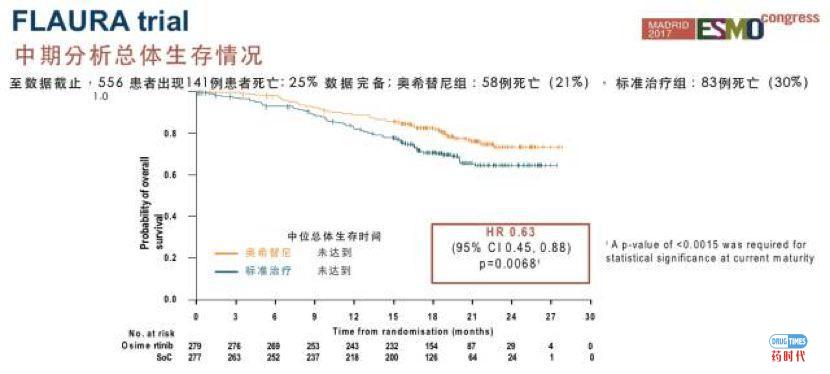
那么,最后还有一个无法规避的问题:奥希替尼虽然能缩小肿瘤,防止进展,但真的能让患者活得更久或者更好么?这也同样是本文所一直在追寻的答案,我们继续来看FLAURA研究,最终评判指标-总生存期(OS)数据目前还不成熟。
截止目前只有25%的患者达到了研究者设计的OS终点,剩余患者还需要进一步收集数据,才能对总生存期进行分析。但是根据初步观察,奥希替尼组患者是有总生存期获益的(HR=0.63;95%CI:0.45-0.88;P=0.0068),最终OS数据还需要更长时间的追踪才会获知……
另外,2019年6月2日于美国芝加哥举办的2019年美国临床肿瘤学会(ASCO)年会上,为了更好地了解奥希替尼在试验随访期的长期生存潜力,研究者使用了数学参数生存模型对FLAURA研究OS数据进行预测,奥希替尼组3年及5年生存率分别为57.3% 和31.1%,相比之下,1代EGFR-TKI吉非替尼/厄洛替尼组3年及5年生存率分别为41.1% 和15.5%。从模型预测的数据来看,奥希替尼一线治疗患者的5年生存率提高了2倍,是非常令人鼓舞的。研究者认为随着未来FLAURA研究的随访数据(OS成熟度为60%)能进一步验证这一发现。据悉,FLAURA研究最终OS的数据将在今年ESMO大会上揭晓。
假设上述试验中的研究终点不采用替代指标ORR或PFS,而是采用患者最终的生存获益指标OS,那么奥希替尼必然不会成为FDA和CFDA历史上最快批准上市的的药物,也无从判断奥希替尼何时可以获批上市,这也就意味着然会有很多患者因为无药可治而提前结束生命,那不是我们想看到的。作为科学家和药品监管机构,是希望通过开发和合理使用新药,让肺癌患者活得更久,活得更好,最终,把这种肿瘤变成可控慢性病。
因此,加速批准是从诊治患者角度出发而应用的途径,进一步要求核实临床效果则是为保证患者长远和得到有效治疗的利益。

以上文字由Biotech venture capital原创,仅为促进讨论与交流,不构成法律意见或咨询建议。版权所有,违者必究。如需转载,请注明作者和Biotech venture capital原创。
参考:
1. 2018 Clinical Trial Endpoints for the Approval of Cancer Drugs and Biologics Guidance for Industry.
2.Surrogate End Points and Their Validation in Oncology Clinical Trials.
3.Randomized controlled trials in the era of molecular oncology: methodology, biomarkers, and end points.
4.Surrogate endpoints in oncology: when are they acceptable for regulatory and clinical decisions, and are they currently overused?
5. A systematic review of trial-level meta-analyses measuring the strength of association between surrogate end-points and overall survival in oncology.
6. Evaluating Progression-Free Survival as a Surrogate Outcome for Health-Related Quality of Life in Oncology— A Systematic Review and Quantitative Analysis.
7. A 25-Year Experience of US Food and Drug Administration Accelerated Approval of Malignant Hematology and Oncology Drugs and Biologics.
过往文章:
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 
