


2023年8月2日,中国科学院上海药物研究所徐华强研究员、段佳研究员和杨德华研究员共同在Nature杂志上发表了最新研究成果“GPCR activation and GRK2 assembly by a biased intracellular agonist”,在国际上报道了第一个高分辨率GPCR——神经降压素受体(neurotensin receptor 1,NTSR1)与GRK2的复合物结构,揭示了GRK2识别和调控GPCR的详细分子机制,并通过结构解析,首次发现了一个全新的GPCR偏向性配体结合口袋,为临床开发靶向GPCR的偏向性药物分子开辟了全新的思路和途径。
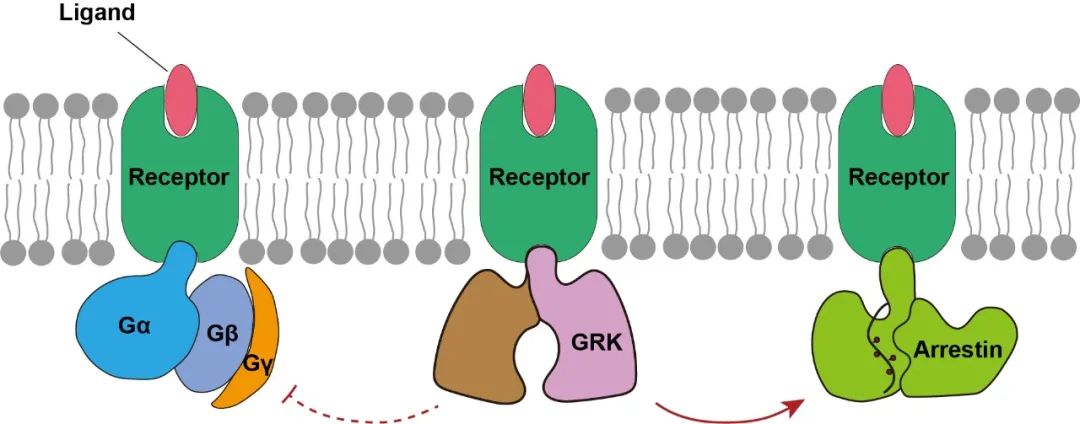

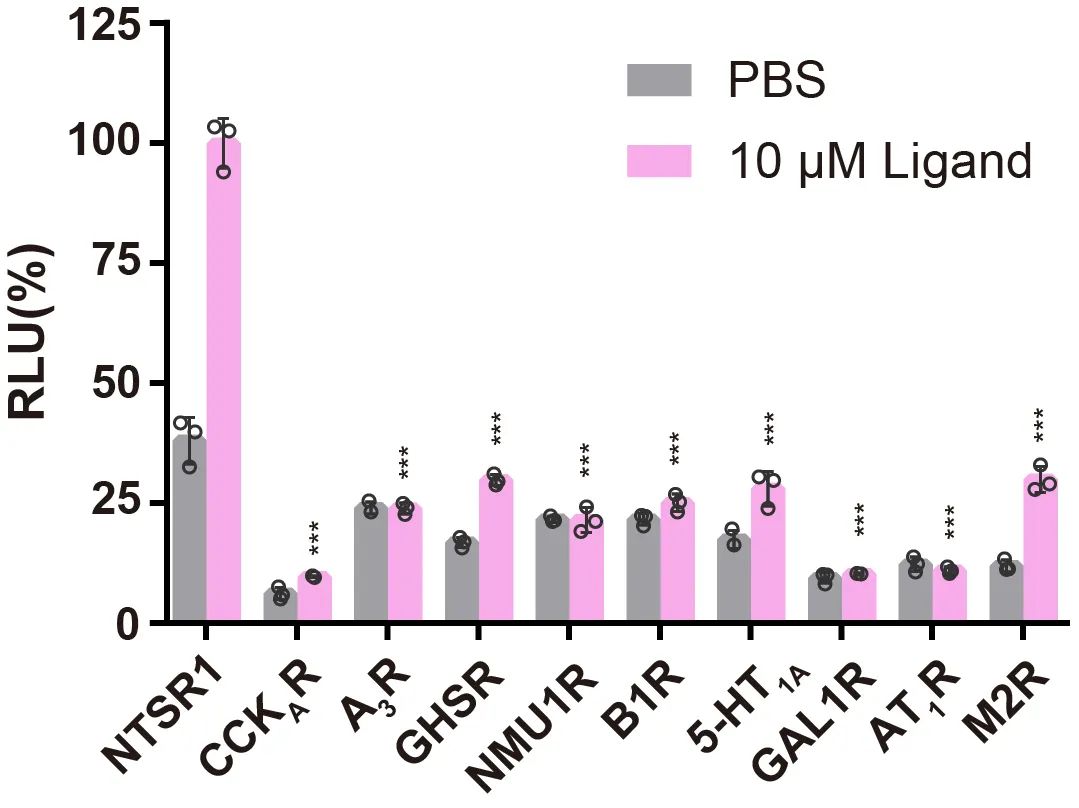
图2 Tango assay的部分筛选结果。所有受体均使用内源性配体激活或者使用PBS作为对照,配体的使用浓度为10 μM,从图上可以看出NTSR1被激活后与GRK2的结合信号最强。
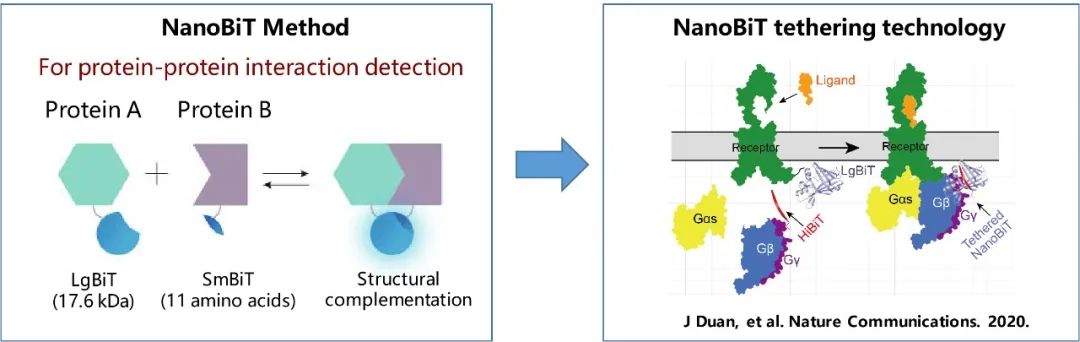
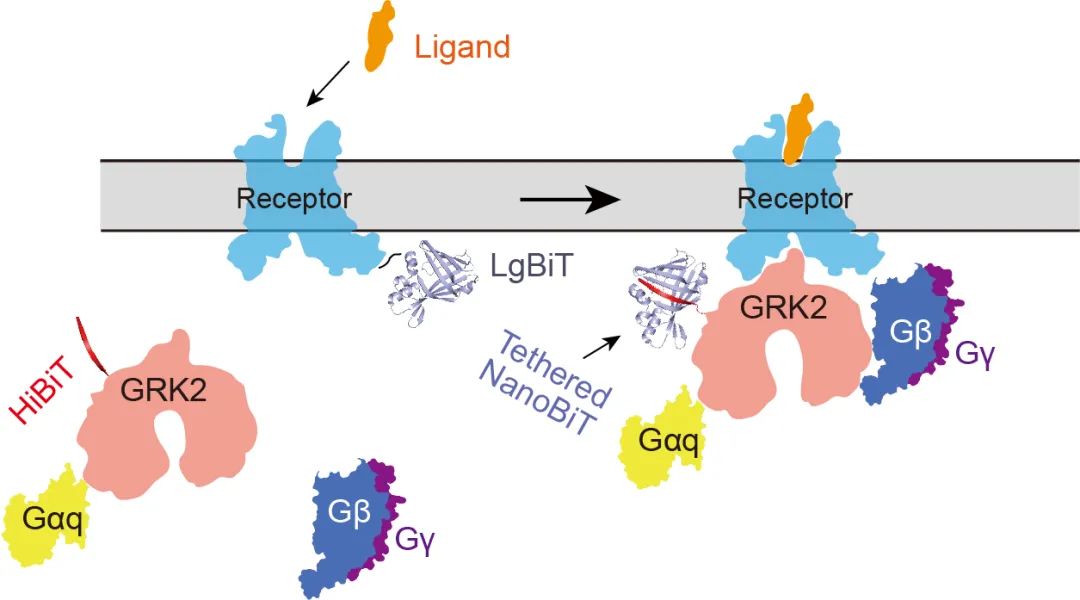
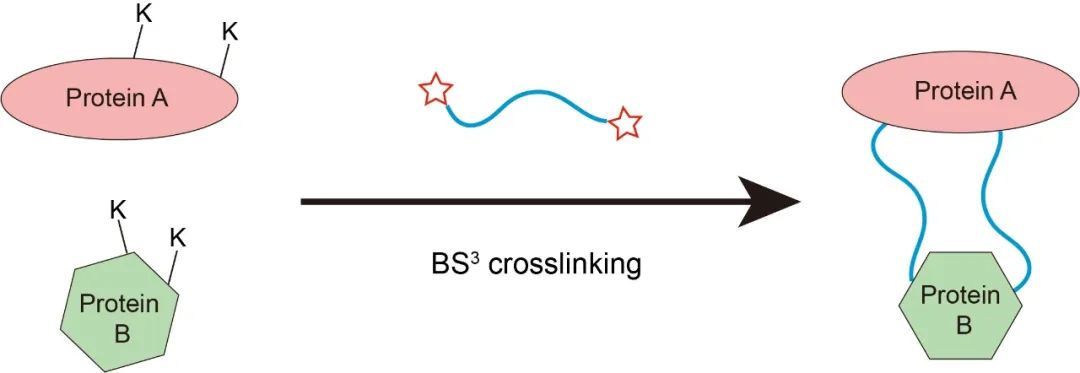
4)采用化学交联技术。为进一步稳定复合物构象,段佳尝试对复合物蛋白进行化学交联。通过文献调研,以及分析受体NTSR1和GRK2的结构特征,段佳猜测受体和GRK的作用界面之间存在较多的赖氨酸,因此重点考虑基于K-K的交联方式。进一步的筛选作者确定了BS3是最合适的交联剂,并对复合物进行了有效的化学交联。
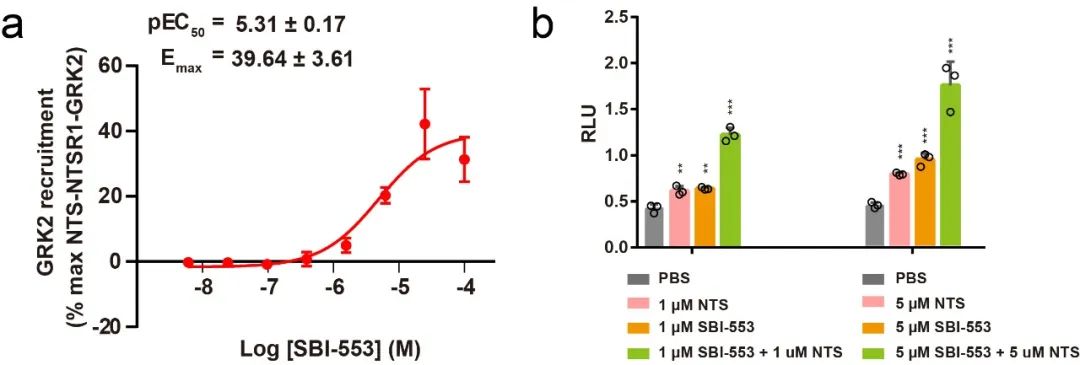
通过结合上述一系列技术手段,作者最终成功解析了第一个高分辨率GPCR与GRK2的复合物结构。结合细胞水平的突变实验,本研究首次阐明了GRK2识别和调控NTSR1的详细分子机制,考虑到GPCRs结构的相似性和保守性,该机制同样可拓展于其他GPCRs。通过结构解析,本研究也首次揭示了SBI-553作用于NTSR1的受体口袋,并发现了SBI-553通过直接占据G蛋白的结合位置从而实现了arrestin偏向性激动的分子基础。
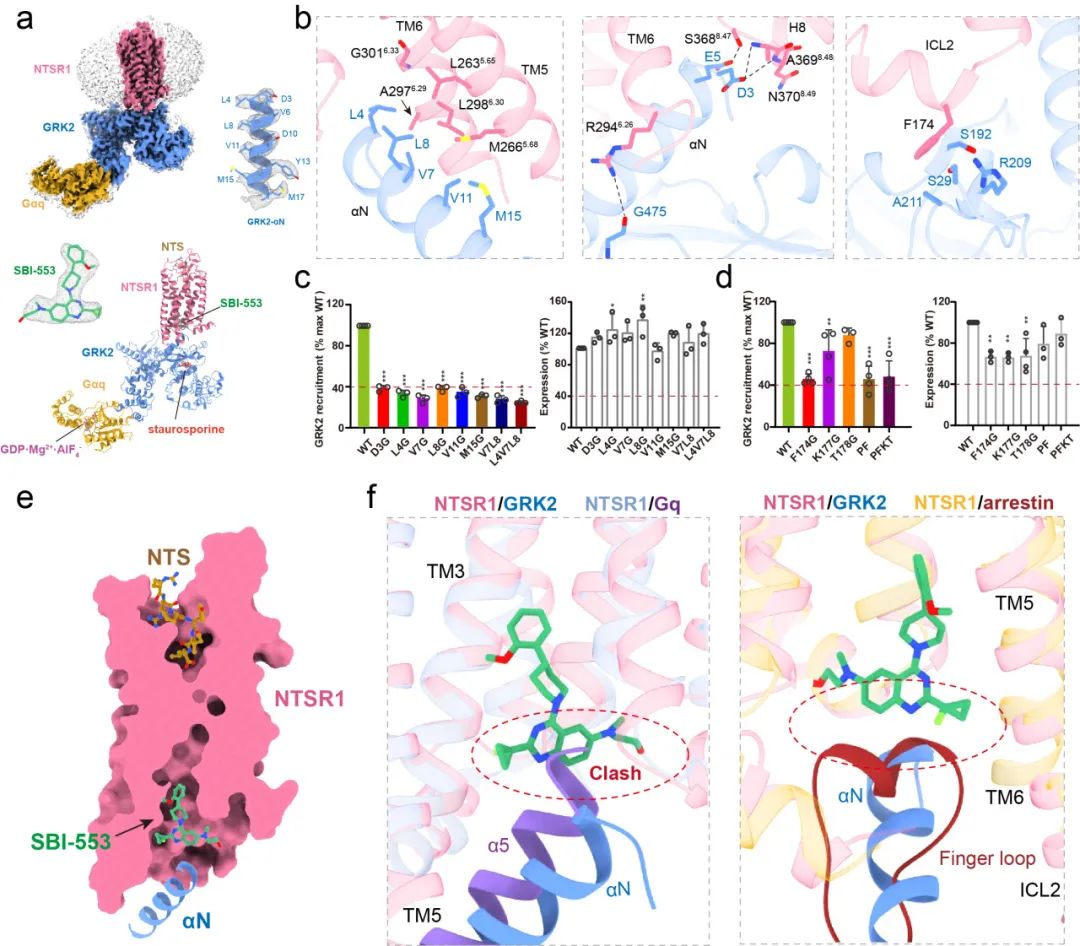
徐华强课题组长期专注于生物大分子复合物的结构解析及功能研究,特别是在GPCRs信号转导复合物的研究中积累了大量经验,本项研究正是在课题组十年研究基础上,通过进一步发展NanoBiT交联技术及化学交联等一系列手段和技术,首次成功解析了高分辨率的GPCR-GRK复合物结构,揭示了GPCR信号转导领域现存的一个最为关键的科学问题,即GPCRs如何受GRKs识别和调控,这一成果是细胞信号转导里程碑式的成果。
GPCRs与GRKs的微弱相互作用和高度动态使得传统的X-射线晶体学无法实现结构解析,本项研究中使用的多种技术方法结合单颗粒冷冻电镜技术将为今后研究其他生物多元动态复合物提供重要的启发和指导依据,从而推动整个生物大分子多元动态复合物研究的进程。
令人意外的是,本项研究中首次揭示了SBI-553结合于受体胞内侧的疏水口袋中,与此前所报道的所有小分子结合口袋均不同,这为今后开发靶向GPCRs的新型偏向性药物开辟了全新的思路和途径。本研究也首次阐明了SBI-553实现偏向性激活受体arrestin通路的结构机制。值得一提的是,徐华强课题组7月31日在Nature上报道了B类GPCRs的新型G蛋白偏向性小分子激动剂,揭示了G蛋白偏向性配体的作用机制,结合本项研究,两项研究首次全面系统地揭示了两种不同类型的偏向性激动剂介导受体偏向性信号转导的详细分子机制,极大地促进了我们对GPCRs偏向性信号转导的理解与认识,为今后开发靶向GPCRs的偏向性药物分子提供了夯实的结构基础。
上海药物所段佳研究员、刘恒博士、赵丰辉博士为本论文共同第一作者。上海药物所徐华强研究员、段佳研究员和杨德华研究员为共同通讯作者。上海市高峰电镜中心执行主任袁青宁对数据的收集及处理提供了大量帮助。该工作得到了国家重点研发计划、上海市市级科技重大专项、中国科学院战略性先导科技专项、国家自然科学基金委等项目的资助。
全文链接
全文链接:https://www.nature.com/articles/s41586-023-06395-9(也可后台留邮箱,获取PDF版本)
专家点评
参考文献:
1、Chen, Q., Plasencia, M., Li, Z., Mukherjee, S., Patra, D., Chen, C.L., Klose, T., Yao, X.Q., Kossiakoff, A.A., Chang, L., et al. (2021). Structures of rhodopsin in complex with G-protein-coupled receptor kinase 1. Nature 595, 600-605.
2、Duan, J., Shen, D.D., Zhou, X.E., Bi, P., Liu, Q.F., Tan, Y.X., Zhuang, Y.W., Zhang, H.B., Xu, P.Y., Huang, S.J., et al. (2020). Cryo-EM structure of an activated VIP1 receptor-G protein complex revealed by a NanoBiT tethering strategy. Nat Commun 11, 4121.
3、He, Y., Gao, X., Goswami, D., Hou, L., Pal, K., Yin, Y., Zhao, G., Ernst, O.P., Griffin, P., Melcher, K., et al. (2017). Molecular assembly of rhodopsin with G protein-coupled receptor kinases. Cell Res 27, 728-747.
4、Kang, Y., Zhou, X.E., Gao, X., He, Y., Liu, W., Ishchenko, A., Barty, A., White, T.A., Yefanov, O., Han, G.W., et al. (2015). Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561-567.
5、Komolov, K.E., Du, Y., Duc, N.M., Betz, R.M., Rodrigues, J., Leib, R.D., Patra, D., Skiniotis, G., Adams, C.M., Dror, R.O., et al. (2017). Structural and Functional Analysis of a beta2-Adrenergic Receptor Complex with GRK5. Cell 169, 407-421 e416.
6、Rasmussen, S.G., DeVree, B.T., Zou, Y., Kruse, A.C., Chung, K.Y., Kobilka, T.S., Thian, F.S., Chae, P.S., Pardon, E., Calinski, D., et al. (2011). Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549-555.
7、Slosky, L.M., Bai, Y., Toth, K., Ray, C., Rochelle, L.K., Badea, A., Chandrasekhar, R., Pogorelov, V.M., Abraham, D.M., Atluri, N., et al. (2020). beta-Arrestin-Biased Allosteric Modulator of NTSR1 Selectively Attenuates Addictive Behaviors. Cell 181, 1364-1379 e1314.
8、Zhou, X.E., He, Y., de Waal, P.W., Gao, X., Kang, Y., Van Eps, N., Yin, Y., Pal, K., Goswami, D., White, T.A., et al. (2017). Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell 170, 457-469 e413.
本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 