IND,Investigational New Drug,一般是指尚未经过上市审批,正在进行各阶段临床试验的新药。IND主要目的是提供足够信息来证明药品在人体进行试验是安全的,以及证明针对研究目的的临床方案设计是合理的。IND申请主要包括Ⅰ、Ⅱ、Ⅲ期临床试验申请,其中Ⅰ、Ⅱ期临床试验为初期临床试验,是疗效的探索阶段;Ⅲ期临床试验为扩大临床试验,是疗效的验证性阶段,只有在初期临床试验IND获准后,申请人才可以提交扩大临床试验申请。中国和美国的IND申请资料通常包括CMC(Chemical,Manufacturing and Control)资料、药理、药代、安全性评估等非临床性资料以及临床资料。其中值得提及的是从药物的研发开始直到药物最终上市,甚至是上市之后,药物的CMC信息始终伴随着药物开发持续更新,且经常在药物申报的文件中,CMC的部分会常常被冠名“活着的文件”。2017年6月19日,中国以第八个监管机构成员国(Regulatory Member)的身份加入了ICH,创新药物研发要求逐步与国际接轨。尤其在对于IND申请中CMC部分,中国NMPA和美国FDA有着大致相同的审核方向,也有一些不同的审核要求。本文就带领大家一起探讨分析IND申报CMC资料要点以及中美差异。
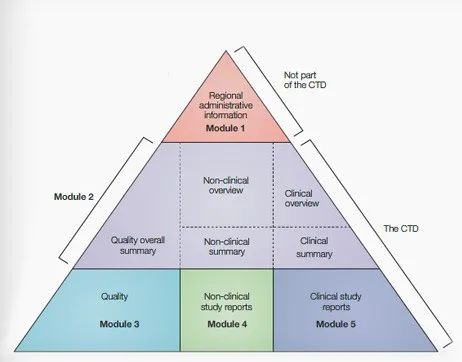
ICH CTD通用技术文件图示CTD是国际公认的药品申报资料编写格式,共分为五个模块:模块一为行政管理文件和药品信息;模块二为通用技术文档总结;模块三为质量;模块四为非临床试验报告;模块五为临床研究报告。其中,模块一为区域性要求,具体内容和格式由相应的监管机构规定,模块二、三、四和五为国际通用要求。CMC的内容主要在模块三。模块三细分为4个部分,包括原料药(3.2.S)和制剂(3.2.P)2个主要部分及其附录(3.2.A)和区域性信息 (3.2.R)。下表描述了IND申请时模块3中原料药和制剂中所包含的内容要点。
总之,对于小分子药物而言,中国NMPA对IND申请中CMC部分的资料的要求更为严格,对于生产过程,杂质控制等方面有着更为细致的规定。对于大分子生物药,中美两国的要求基本一致。无论怎样,对于企业而言,IND申报仍然值得关注的重要课题。CMC部分包含很多复杂的技术信息,并且药政指南详细且繁琐。申报时选择有经验的第三方顾问团队或为不错的选择。参考资料: [1]. ICH M4Q(R1): CTD on Quality. September 2002[2]. ICH Q1A(R2) Stability Testing of New Drug Substances and Products. August 2003[3]. ICH Q3A(R2) Impurities in New Drug Substances. October 2006[4]. ICH Q3B(R) Impurities in New Drug Products. June 2006[5]. ICH Q3C(R8) Impurities: Guideline for Residual Solvents, May 20, 2021[6]. ICH Q11 Development and Manufacture of Drug Substances. November 2012关注“药时代”微信公众号,获取更多精彩内容 ↓ ↓ ↓








 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 