关注“药时代”微信公众号,获取更多精彩内容 ↓ ↓ ↓


前后20天里,关注中国新药出海的人,心情如过山车一般跌宕,久久不能平息。而临床试验这个开发药品的基础环节,也成为大家热议的话题。
2月26日药时代、国际临研、昆翎医药合办了一场线上活动,主题为:破解申请FDA上市批准从「起初失败」到「最终获批」的密码。并邀请了张丹博士和罗晟教授进行了分享。
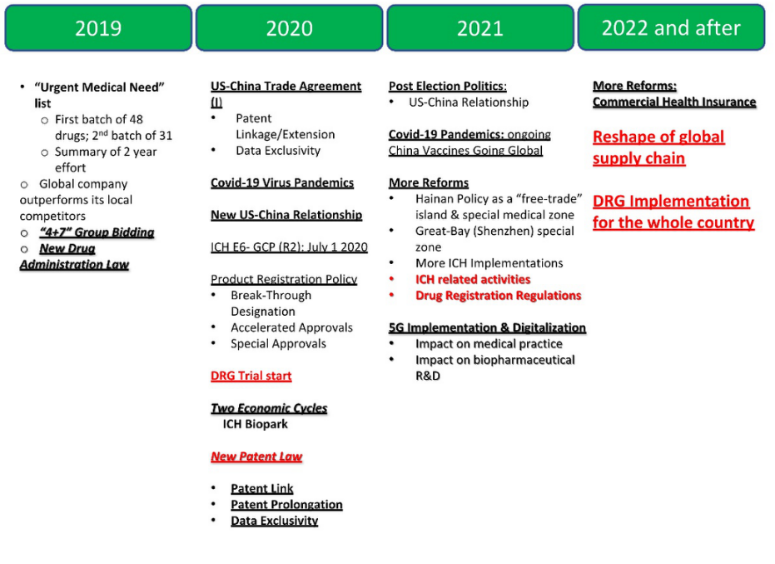
会上,张丹博士从个人经历出发,讲述了中国创新药环境近年来的变化,主要是2017年中国加入ICH,以及2020年之后的后疫情时代,分别影响了中国引进国际创新药的速度,与“从IND到NDA”的速度。
后疫情时代,药品从IND到NDA的时间,从之前的6~7年变为了平均7个月,大大影响了药企对NDA的规划。
随后,张丹博士分别介绍了百济神州的成功案例,和早年他自己亲身经历的两次NDA申报经历。
其中一场,是一款针对肾透析病人的产品,在与FDA沟通临床开发路径遭到FDA拒绝后,仅仅通过改变了临床指征,就利用早期19例病人惊人的临床数据,获得了FDA的NDA批准,非常具有参考意义。
药时代对张丹博士的演讲内容进行了整理,下拉至文末观看完整视频,需要张丹博士演讲PPT的朋友们请在文章留言处留下您的邮箱(尽量不要在后台留言喔,方便我们统计,避免遗漏)。
首先感谢药时代能提供这样一个平台和机会,跟大家分享一些心得体会。
今天主要结合我公司或个人在美国申请NDA的一些体会,对比中国申请NDA的一些考虑,谈谈从国内企业的角度看,如何申报美国NDA。
2015年,毕井泉成为了国家药监局一把手,自此迅速开启了中国药监系统的改革,其中涉及到了很多影响我们现在NDA的申报过程。记得在ODAC会议上,FDA曾提出:中国80%的临床试验数据不可靠,这起于2015年“722”事件,当时国内企业撤回了83%的NDA申请。
2016年,国家重新定义了“新药”的概念,认为:“全球新”在中国才是新药,推翻了此前的定义。
2017年,国家药监系统作为监管成员加入了ICH,这是具有里程碑意义的。中国加入ICH后,第一个迅速接受的指南就是ICHE17(多国多中心临床试验指南),也是这次ODAC会议反复强调的。
2018年,国家开始大批量接受海外数据,大大加快了国际创新药进入中国的速度。
2019年,国家出台了“集采政策”,同时为了适应ICH的加入,重新修订了《中华人民共和国药品管理法》。
2020年,又有了新的举动。1月份跟美国签署了《中美贸易合作协议》的第一部分,其中专门有针对创新药的条款,主要是专利链接、专利补偿、数据保护等。因为这个条约,国家在2020年年底,把《专利法》中涉及药物的部分进行了修改。
2021年,出现了Covid-19事件,在这期间,药品从拿到IND到获得上市批准的时间(EUA批准、有条件批准国内的特殊批准等),由原来的6~7年变成了7个月,无论是疫苗还是治疗性药物都变了。新疫情时代,我们对于NDA的战略、NDA的考虑、临床的规划都将发生翻天覆地的变化。
记得在ODAC会议前期乃至当天,美国癌症审评卓越中心的主任和他的同事专门谈到了,“我们希望临床试验是MRCT(国际多中心)”。为什么?原因之一是在疫情时,多国、多中心的临床试验能够加速NDA批准,因为入组快了。而且国内药企在疫情期间,做的也是多国多中心,因为中国疫情的处理太成功,国内没有足够的病人支持任何大规模的临床研究,我们的疫苗甚至是治疗药物,包括中和抗体、开拓药业的雄激素受体药等,都是做的国际多中心。
因此,疫情使得大家不得不“国际多中心”,而且凑巧在疫情爆发前的1、2年,ICH通过并且在全球推广了E17多国多中心临床试验指南。可以说,无论在理论基础、监管的趋同、还是现实条件,都逼着大家走国际多中心,这只有通过这条路才能加速IND到NDA的过程。
从这个角度讲,这次ODAC会议只是阐明了一个事实:不走MRCT,你很可能会耽误在某个国家甚至多个国家未来的“NDA加速战略”。
因此,出于几个方面的原因:中国医保政策连续不断的变化、中国加入ICH、创新药内卷等等,国内企业如果不做MRCT、不考虑多国的NDA申报,而是只在某一个国家甚至只做国内市场,将无法维持长期的高强度、高成本、高风险的研发活动。
必须找到高回报的地区进行市场化,包括美国、欧盟、日本,甚至“新丝绸之路”国家,只要有足够的市场。疫苗就是典型的成功案例,去年有20亿剂疫苗卖向“新丝绸之路”国家。NDA的策略关系到公司的生存发展,不得不现在就考虑好,而且必须是多国、多中心。
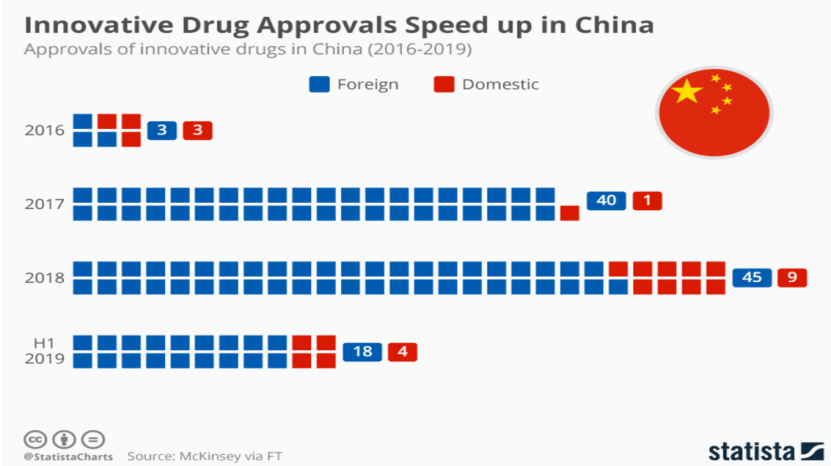
下图是2016年~2019年H1期间,中国批准的创新药。
2016年,中国还没有加入ICH,国家批了6个新药,其中3个国产、3个进口;
2017年、2018年、2019年,批准的创新药主要是进口,因为数据互认了。
疫情期间,国内企业追赶上来,2021年批准的新药,一半以上来自国内,但问题是所批的药中绝大部分是“me too”,有时甚至是“we too”,形成了内卷。
因此,我们要走向国际。无论这次ODAC会议怎么讲,那只是暂时的一过性现象,并不阻碍大家寻找“未满足的临床需求”,无论是美国的、欧洲的、日本的、“丝绸之路”国家,还是中国的“未满足需求”。
最成功的的例子就是百济神州,他们通过开发孤儿药指症,解决了一个未满足的临床需求,其中二期的关键临床数据全部来自于中国,当然它一期有国际数据(第一次人体试验是在澳大利亚,药物药物相互作用、药物实物相互作用、心脏毒性等是在美国做的),牵头的医生、PI是北京肿瘤医院的医生,通过FDA现场稽查获得批准。
只要找到了快速获批的路径——孤儿药,美国就有批准的NDA的灵活性,百济神州这个药享受了breakthrough、Fast Track、Accelerated Approval、Orphan indication,几乎所有的加速因素都拿到了,而且完美的利用ICH,把美国的监管条件和中国的大量病人有机的结合在一起,临床一期全球化(澳大利亚、美国、中国),关键性的二期临床数据则全部来自中国。
所以,这次ODAC会议虽然没有立刻批准单一中国数据申报NDA,但也并没有拒绝这个产品将来再申报,甚至改换指征再申报。目前,在中国的内卷、中国医保环境的改变、ICH环境下的全球竞争,以及疫情带来的新局面下,坚持国际化,坚持多中心,是我们企业在全球寻找突破的必由之路。美国这些加速因素是多年形成,大家可以认真领会。百济神州做了一个很好的例子,这是中国作为创新药成药第一次在美国获得上市的批准,所以,成功的先例就在前面。
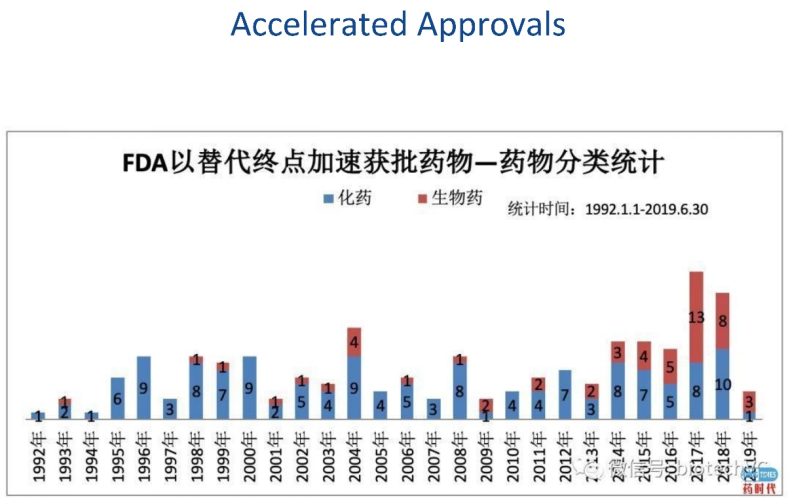
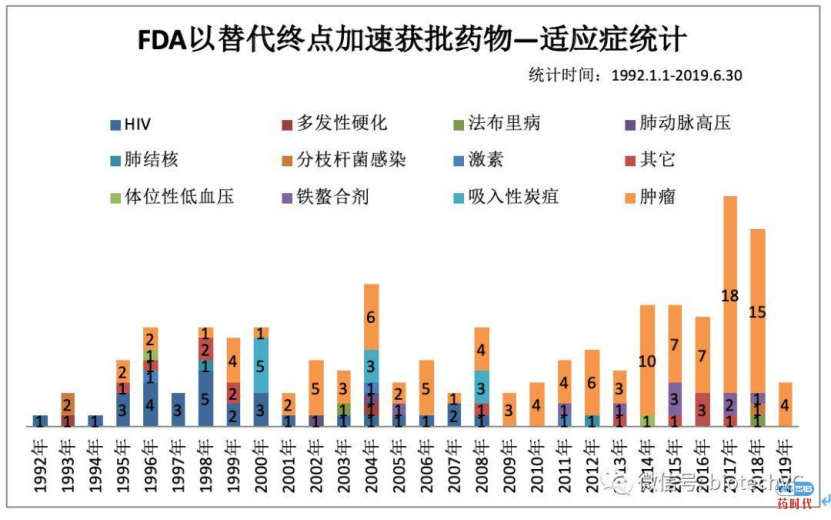
FDA为了加速批准,某些时候同意替代终点。下图是每年用替代终点批准的药物,可以看出来替代终点的使用也是越来越多。
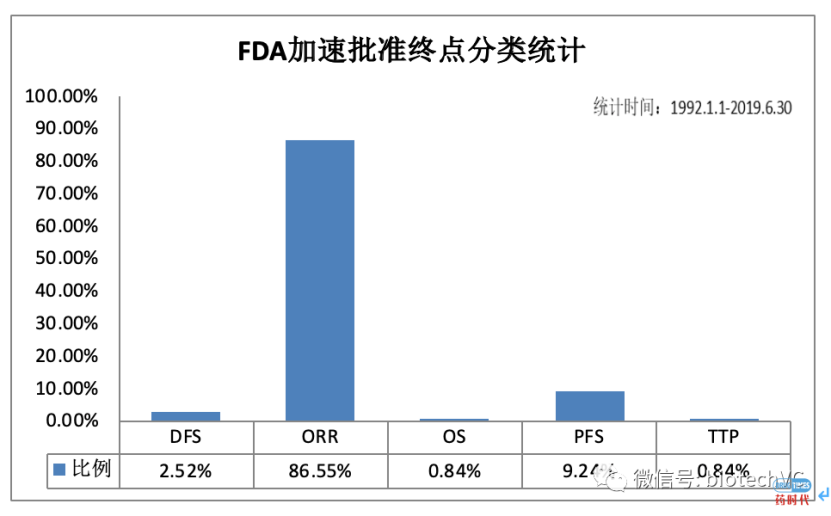
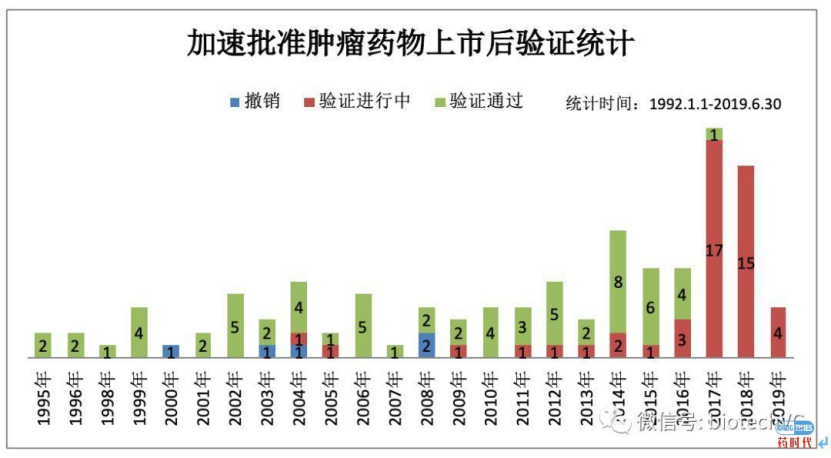
下图是FDA使用加速批准的替代终点分类,使用最多的是ORR和PFS。下图不同的治疗领域中用替代终点获得NDA批准的例子,特别是肿瘤药物,加速批准的种类特别多。
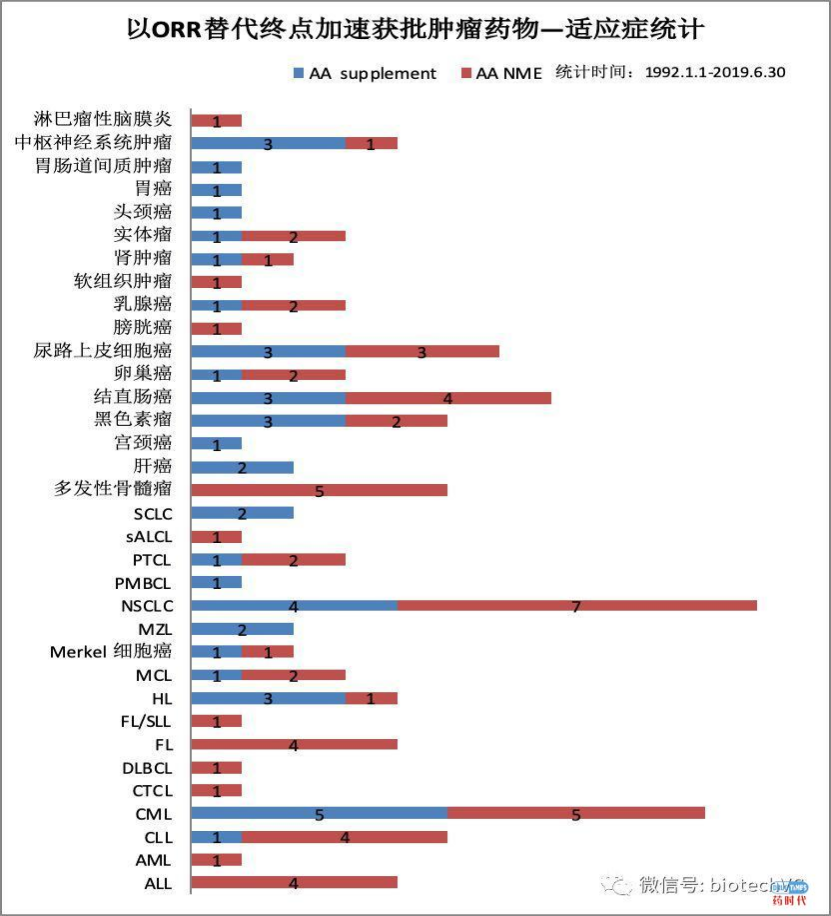
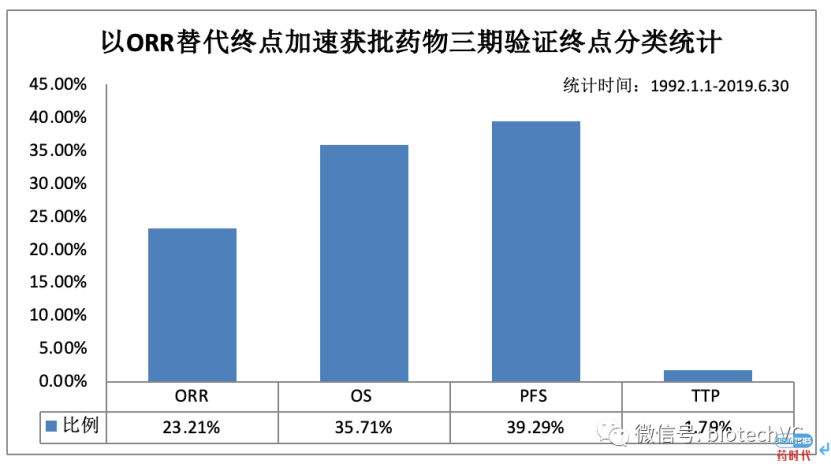
这里有更详细的,介绍哪个肿瘤适应症是用ORR替代终点获得美国批准的。以及以ORR在关键性三期验证临床试验中的使用比例。下面给大家罗列一些,在美国批准NDA做的临床试验一般情况,总的来讲,你要做Orphan indication的话,需要的总例数远小于非Orphan drug的临床试验的总数。下图是在美国批准NDA需要做的临床例数分布图。凡是小于500的,特别小于200病人的,差不多都是Orphan indication。美国启动了一个project orbis,把加拿大、澳大利亚、瑞士、新加坡、英国、新西兰、巴西纳入了其中。这些国家不光是数据互认,并且同时审批NDA/BLA肿瘤产品。这次ODAC会议之前,Richard Pazdur美国肿瘤审评卓越中心主任,提议中国是否也考虑加入project orbis,同时审批NDA,几乎同时上市。这是另一条可能的加速的路径。百济神州的泽布替尼进入美国,和记黄埔的呋喹替尼也是主要利用中国的临床数据。无论你怎么样,都要充分利用ICH各种指南,特别是正在开发的ICH Adaptive Design(E20)指南,采用Adaptive Design确实能够比较显著增加你在美国NDA批准的成功率,有人预估能提高13%的NDA成功率。第二类是以病人为中心的临床试验设计,也能提高美国NDA的成功率。以biomarker为基础的精准医学临床设计,也会增加NDA成功率。以适应真实世界的研究数据为基础的临床试验设计,也会显著增加美国NDA成功率。这些最新的理念,虽然还没有发表的ICH,应该予以早期的跟踪,美国已经有了Adaptive Design这些指南等等,在我们设计多中心临床试验时都要把这些最先进的理念用上。特别是在疫情期间,美国和WHO所设计的国际多中心大规模的研究,几乎都是Adaptive Design。最后,分享一些NDA成功与失败案例,这些是我个人亲身经历的。我以前在一个欧洲公司负责它北美的临床医学,第一个申请的是间歇性跛行的indication,做了2个关键性III期的国际多中心临床试验,第一个是北美临床试验做了1400人,结果是我们的Primary Endpoint用了maximum walking distance(最长步行距离),这是美国FDA认可的金指标。北美临床研究P=0.052,欧洲临床研究(东欧+西欧)P=0.02,按着规定,必须做2个关键性临床研究,把两个数据荟萃分析得出P=0.04。FDA专门挑选了我们的一个在的欧洲中心进行详细的现场稽查,去的前苏联巴甫洛夫研究所(牵头单位),做了详细的调查,没有发现任何问题。虽然我们通过了现场稽查,FDA最后依然没有同意NDA申请,要求我们再做一个临床试验。为什么?因为我们事先没有经过FDA同意,可以用荟萃分析的结果作为NDA的关键性结果,在FDA眼里,他们同意的分析方案是两个独立的关键性研究,所以一个阳性、一个阴性,特别是北美的阴性结果,让我们没获批,不得不再做下一个III期临床研究。这是一个活生生的例子。同样一个公司,我们也有反败为胜的例子。这是我们在美国做的,只做了19个肾透析的病人临床试验。当初是我们希望拿这个作为一个临床试验的探索,再做一个更大的以肾透析病人的临床症状改善为指征的NDA。结果跟FDA开会,FDA完全不同意我们的临床开发论据,我们的NDA看起来前途渺茫,没戏。但很快我们内部在专家的提醒和内部联合讨论下,我们就用这19例病人,改变indication,就提出来你就批准我们做肾透析病人的carnitine deficiency这个治疗指征。肾透析的病人会大量丢失肉毒碱,肉毒碱是三羧酸循环中的关键辅酶,19个病人明显比正常人carnitine的含量大幅度改变,就拿这19个病人的数据,通过改变指征,就获得了美国FDA的NDA批准。所以在申报NDA的路上,即使产品第一次遇到了障碍,但并不妨碍我们利用这个产品特有的优势,在多个指征、多个审批路径上重新考虑,获得成功。如果您需要张丹博士的PPT进行学习,在文末留下邮箱地址,我们会在汇总后统一发送。
药时代近期举办的直播活动受到了朋友们的广泛关注和支持,为了给大家带来更高质量的内容,我们也在不断更新拓展资源。
好消息是,近期国际顶级医学期刊《新英格兰杂志》将举办一场会议,邀请我国的明星药企高管+名校教授+顶级临床PI一同参与讨论,产、学、研、医相结合,质量非常高,具有很强的参考价值和实际意义。
药时代也受邀作为媒体平台支持,将全程直播本次重磅会议,点击下方预约,与药时代一同学习!
关注“药时代”微信公众号,获取更多精彩内容 ↓ ↓ ↓
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!
 点击这里,观看全场视频回放!
点击这里,观看全场视频回放!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权







 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 