



为指导和规范基因治疗产品的临床试验,2021年12月3日,国家药品监督管理局药品审评中心官网发布《基因治疗产品长期随访临床研究技术指导原则》(成文时间2021年12月1日),自发布之日起实施,本指导原则针对基因治疗长期随访临床研究的观察方法和研究设计进行讨论,着重阐述了基因治疗长期随访临床研究的观察目的、考虑要素、设计实施以及不同基因治疗产品的特殊考虑等相关要求,在为该类产品开展长期随访临床研究提供技术指导,确保及时收集迟发性不良反应的信号,识别并降低这类风险,同时获取这类产品长期安全性和有效性的信息。

本文为大家梳理了基因治疗产品长期随访临床研究内容并结合自己的解读与大家分享。
尽管基因疗法在治愈疾病方面具有极大潜力,但它也会对人体带来长期或永久性影响,接受基因治疗的患者出现迟发性不良反应的风险可能会增加,如基因治疗产品的活细胞的生物学特性的变化在体内长期存在,可能增加不可预测的风险。为了评估和降低这类风险,并了解治疗效果随时间延长的变化,有必要对参加基因治疗临床试验的受试者开展长期随访。
目前,美国FDA和欧盟EMA均已发布相关技术指导原则,经查询欧盟EMA于2008年发布的《Guideline On Safety And Efficacy Follow-Up-Risk Management Of Advanced Therapy Medicinal Products》和2009年发布《Guideline On Follow-Up Of Patients Administered With Gene Therapy Medicinal Products》指南,为申办方提供了基于基因治疗的风险概况,考量长期随访的指导性建议。
美国FDA于2020年1月发布的《Long Term Follow-Up After Administration Of Human Gene Therapy Products》,对如何设计长期随访研究提出了建议,并对长期随访观测要素、持续时间、数据收集和报告要求等问题提供了指导思路。
考虑到国内尚无相关指导原则对基因治疗产品长期随访临床试验设计进行规范指导,2019年4月,国家药监局启动了中国药品监管科学行动计划,药审中心负责实施的“细胞和基因治疗产品技术评价与监管体系研究”纳入首批研究项目,其中,《基因治疗产品长期随访临床研究技术指导原则》是基因治疗类药物技术评价体系的重要内容,有助于引导基因治疗类药物临床试验的规范开展,CDE在充分调研国内外同品种研发情况以及相关临床试验技术要求基础上,自2021年1月启动,2021年6月发布征求意见稿(会稿截止时间2021年7月4日),2021年12月3日,CDE正式发布《基因治疗产品长期随访临床研究技术指导原则》,自发布之日起实施。
基因治疗产品长期随访的主要目的是收集受试者的迟发性不良反应,了解基因治疗产品在体内的存续情况,从而识别并降低接受基因治疗产品的患者的长期风险。
1、长期随访的持续时间多长合适?
基因治疗作为一种新兴的疗法,其长期安全性仍是未知的,长期随访的持续时间应确保足以观察到受试者因产品特性、暴露情况(生物分布和给药途径)等导致的风险,应不短于迟发性不良反应的预期发生时间。一般而言,针对不同类型的基因治疗产品长期随访观察时间建议如下表:
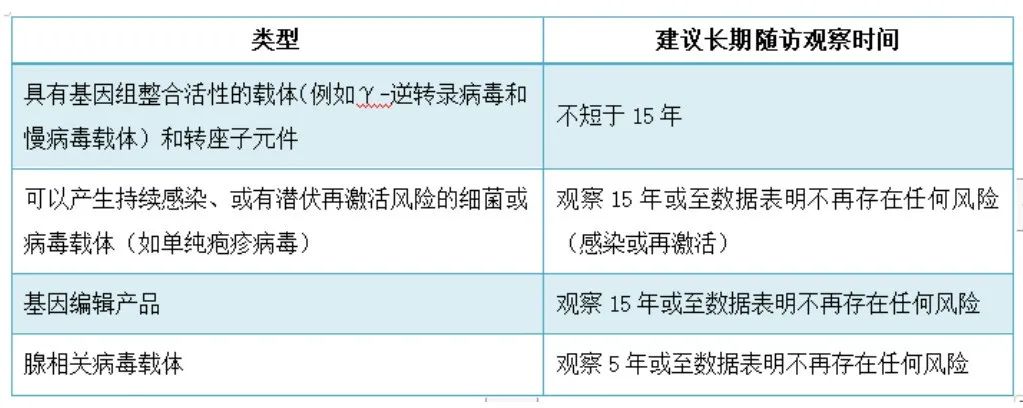
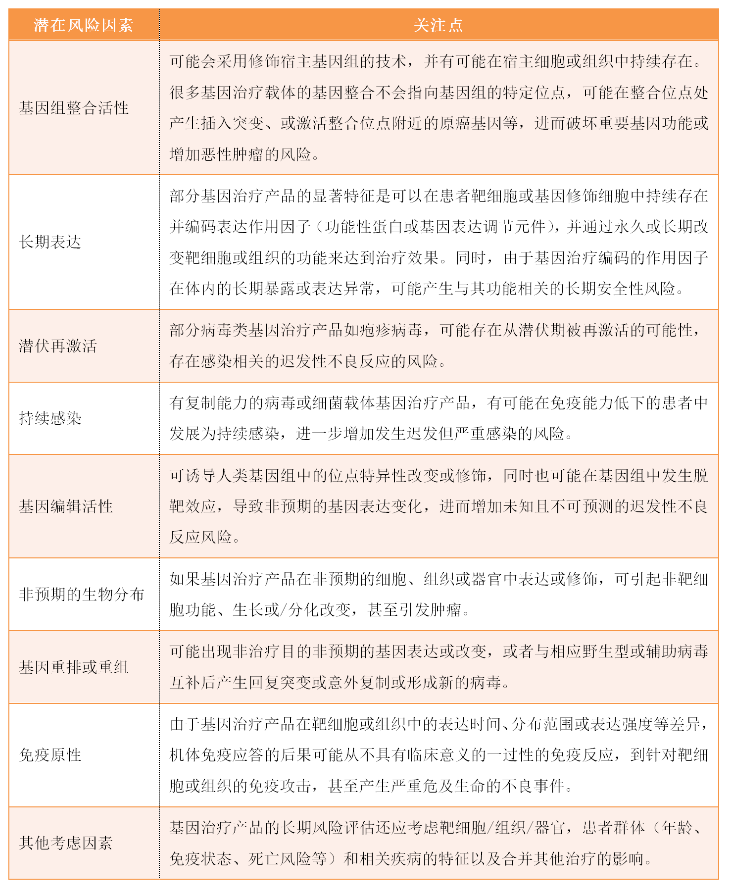
2、迟发性不良反应相关的潜在风险因素评估
在评估基因治疗产品的风险因素时,申请人应考虑基因治疗产品的特性,同时参考该产品的非临床和临床数据以及类似产品的已知数据,申请人应尽可能在非临床研究中获得用于评估迟发性不良反应风险的数据。包括但不限于以下几个要素:
3、临床研究人群相关的潜在风险因素评估
(1) 在设计长期随访临床研究的方案时,应考虑目标受试者人群及特征、整体健康情况以及接受治疗的患者的预期生存期等特征对迟发性不良反应的收集的影响。
(2) 当临床研究人群的某些特征(如预期寿命短、多重合并症、以及暴露于放疗或化疗等其他药物)可能干扰迟发性不良反应的观察分析时,会影响长期随访观察在评估和减轻受试者风险方面的效用;
(3) 而在病情较轻或较局限,合并症以及伴随治疗有限或较稳定的受试者中,通过长期随访观察收集到的评估数据可能更容易分析。
4、长期随访的设计实施
(1) 知情同意
1) 内容:需包含长期随访研究的目的、研究程序、持续时间、访视间隔以及研究者、伦理委员会或申办方的联系方式等。
2) 注意事项1:当非临床研究或临床试验中发现基因治疗产品的风险有所改变时,应及时更新知情同意书并告知受试者。
3) 注意事项2:知情同意书中还应对长期随访期间的人体组织样本采集和保存、基因检测等进行说明。
(2) 长期随访临床研究方案设计
1) 内容:应详细说明受试者的监测计划,包括访视时间表、采样计划、监测检查方法以及长期随访临床研究中的目标临床事件等。
2) 注意事项1:建议申办方提供一份简明科学的随访记录指导,供研究者及相关医务人员(包括研究者以外的医生和护士)记录所有观察结果和与研究相关的所有数据。如果在临床试验期间或上市后获得改变基因治疗产品风险的重要信息,应及时修订随访计划并予以实施。
(3) 长期随访实施
1) 在受试者接受基因治疗后的5年内(或根据具体产品的风险确定的长期随访期内),临床随访应记录受试者的简要病史,使用致癌或致突变药物和其他药物的情况以及有关的不良事件信息,新出现、复发或加重的疾病(例如恶性肿瘤、神经系统疾病、免疫原性或自身免疫类疾病、感染、甚至死亡等)及相关体格和实验室检查、受试者及其配偶的妊娠和生育情况等。同时,尽可能在合适的随访时间点采集相关样本,使用经过验证的、足够敏感的方法检测基因治疗产品在体内的持续存在情况并分析相关影响,直至数据表明不再有任何风险。如随访过程中出现疑似与基因治疗产品相关的不良事件,应及时根据临床、实验室、分子生物学、细胞遗传学、组织学或HLA分析获得的证据或深度测序数据等进行相关性的因果分析,必要时提高随访频率或增加随访内容。
2) 对于随访时间超过5年的基因治疗产品(或根据具体产品的风险确定的长期随访期内),完成前5年的随访后,可通过电话或书面调查问卷等方式,并尽可能采集相关样本,保持每年至少随访受试者一次直至随访期结束。如前期随访提示产品在体内持续存在,建议观察至数据表明不再存在任何风险。
版权声明/免责声明
本文为授权转载文章,仅代表作者观点,版权归作者。
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!

推荐阅读
-
精彩回放!一起回顾2021中国新药CMC高峰论坛次日要点! -
奥默医药宣布提前完成国家“重大新药创制”奥美克松钠首个适应症III期临床研究 -
官宣!74种药品新增进入国家医保目录 -
辉瑞专栏 | 糖衣口服固体制剂的挑战:更易吞服的药丸 -
博志研新GRDDS技术平台取得重要突破,CNS改良新药获得IND受理! -
英矽智能启动 ISM001-055 首次人体试验,快速推进其端到端人工智能平台所发现新药的临床验证 -
进博如高考,这家百年跨国药企“键”入佳境,“中国关键”项目成绩斐然! -
收藏!2021年中国国家药监局(NMPA)“官宣”批准的创新药、疫苗(持续更新中) -
徐亦迅博士 |“诺光灯”下的科学史掠影 (下)——2008年化学奖:生物发光现象与绿色荧光蛋白的发现 -
万字长文!ADC烽烟四起,III期临床群雄逐鹿:老牌玩家的“内”与“外”;HER2靶点之争;新靶点“三剑客”,创新不止于靶点 -
如何创新永续,在雨后春笋般成长的新药公司之林中保持领先地位?| 药时代专访德琪医药创始人、董事长兼CEO梅建明博士 -
今天是感恩节!我们感谢张文宏医生、千千万万的医生朋友们,以及新药和疫苗背后的他们 -
感恩节大礼!NMPA官宣批准两款重磅产品!热烈祝贺亚盛、思路迪/康宁杰瑞/先声药业! -
精彩回放!一起回顾2021中国新药CMC高峰论坛首日要点! -
中国新药研发20年!——20位大咖回顾那激情燃烧的难忘岁月 -
“群贤毕集,创研未来”,2021勃林格殷格翰创新大赛完美收官! -
2021中国新药CMC高峰论坛现场盛况全纪录!6个小视频,1800+照片,12万+浏览,精彩瞬间永留心间! -
《Cell》重磅!免疫记忆可被大脑唤醒!为什么开朗的人不容易患癌?都和大脑调节免疫机制有关 -
收藏!CDE发布《以临床价值为导向的抗肿瘤药物临床研发指导原则》 -
药时代报告 | 谢雨礼博士解读有机化学赋能药物发现的新课题 -
辉瑞专栏 | 成功的CDMO合作如何为无菌制剂产品的成功奠定基础 (第六部分,共计六部分) -
创新致胜的今天,和铂医药的专利库里都有哪些”大杀器”? -
极目生物与温州医科大学附属眼视光医院签署战略合作协议 -
全球临床Ⅲ期及以上小分子口服新冠药盘点 | 新药VS老药——能抓老鼠就是好猫! -
3.92万美元/年,罗氏的阿尔茨海默病新药定价曝光 -
先机勃发 · 共创未来!勃林格殷格翰与国内多家顶级医院在2021进博会签署临床研究框架协议 -
诺和益®进博首秀,1.3亿糖尿病患者新选择!——专访诺和益®中国3期临床研究主要研究者、解放军总医院内分泌科主任母义明教授 -
进博会力促创新合作——勃林格殷格翰与上海之江生物签署新冠双特异性抗体药物CDMO合作协议 -
ADC药物Q3大卖1300万美元,这家biotech股价却大跌 -
揭秘奥默,一家挑战“今日之药”的创新药企,勇气可嘉,实力何在? -
辉瑞新冠口服药物Paxlovid降低住院或死亡风险高达89%!股价大涨! -
里程碑!默沙东口服新冠药物molnupiravir获批! -
三迭纪:创业六载,步履不停,开启3D打印药物的2.0时代 -
伏美替尼一线治疗达到临床终点,EGFR靶点为何经久不衰? -
金嗓子退市风波启示录 | 药企发展动力来自创新 光靠吆喝难以永留市场“芳心” -
5.25亿美元!默沙东布局细胞因子疗法 -
里程碑!PNAS | 人源全长PI3Kα复合物三维结构成功解析(附:邵峰院士、叶德全院长点评) -
药企股价破发频现,为IPO立项的Me Too项目由谁买单? -
重磅!上海打造新“诺贝尔奖”,红杉中国捐赠5亿元独家支持 -
大咖云集!第四届世界顶尖科学家论坛隆重开幕 68位诺奖得主参会 -
中药专家赵军宁任国家药品监督管理局副局长 -
默沙东,一家值得尊敬的公司!将向105个发展中国家分享抗新冠口服药,用行动践行增辉生命的承诺! -
复星医药疫苗业务受制于人 欲40亿深耕疫苗领域 -
11月11日,欢聚张江,共话我和我的CMC!——第二届中国新药CMC高峰论坛期待您的光临! -
2348位同药已投票!关于PD-1和恒瑞的调查结果,现在公布! -
150个小视频,26万+次播放,讲述新药研发奇幻之旅!那些人,那些药,那些感人的故事。。。 -
从鲁迅想到新药研发 | 世上本没有药,吃的人多了,安全有效,也便成了药 -
2021年H1全球前5大糖尿病药企营收及主要产品盘点 -
强生创新网络研讨会 为创新者带来机遇! -
美国CDMO也疯狂!~ 这家“卖水人”一周吞并三家公司 -
前FDA检查官Peter Baker先生诚邀广大朋友们出席2021中国新药CMC高峰论坛! -
视频 | 纪念中国出生的诺贝尔奖得主Edmond Fischer教授! -
陈力博士、华烨博士热烈欢迎朋友们莅临第二届中国新药CMC高峰论坛! -
收藏 | 十大跨国药企的Top3拳头产品盘点 -
谢雨礼博士 | 有机化学又获诺奖,药物创新发动机面临四大新课题(文末有彩蛋!~) -
为中国创新药插上翱翔全球的翅膀!——「创新药全球布局与国际标准生产质量高峰论坛」在张江成功举办! -
破局当下新药研发的困境——大数据与人工智能的多维度利用 -
CDE杨志敏 | 以临床价值为导向的抗肿瘤新药研发 -
上海国际生物医药产业周——张江生命科学国际创新峰会召开,“张江药谷”打造世界级生物医药产业集群 -
不入张江,焉得辉煌?!18位大咖邀您共赴张江生命科学国际创新峰会! -
辉瑞专栏 | 成功的CDMO合作如何为无菌制剂产品的成功奠定基础 (第四部分,共计六部分) -
GLP-1的前世今生——追寻Best-in-Class GLP-1RA的爱恨情仇 -
和铂医药FcRn抗体进入临床三期!有望领跑10.8亿美元中国市场! -
百济神州、辉瑞、BMS的Logo都换了,恒瑞的还远吗?~~(请投下您宝贵的一票!) -
恒瑞国际化迈出新的一步!祝贺Eid博士担任恒瑞(美国、欧洲)首席医学官!他曾在BMS、默沙东、罗氏等跨国药企担任高管 -
默沙东新冠口服药:小概率成功背后的残酷真相 -
默沙东新冠小分子–机理与结构





 点击这里,与同药们在这里喜相逢!
点击这里,与同药们在这里喜相逢!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 