

基因治疗(gene therapy)是指将外源正常基因导入靶细胞,以纠正或补偿基因缺陷和异常引起的疾病,从而达到治疗目的,也就是通过基因转移技术将外源基因插入患者适当的受体细胞中,使外源基因表达的产物能治疗某种疾病。
从广义说,基因治疗还可包括在核酸水平治疗某些疾病的措施和新技术,如RNA干扰技术。基因治疗技术从1990年开始第1例临床试验至今,经历了近30年的发展,其间一度跌入低谷,后在争议中前行,直至最近几年取得了不俗的进展,重新成为各国研究的热点。
2019年5月美国食品药品监督管理局(FDA)批准了1个用于治疗小儿脊髓性肌萎缩症(SMA)的基因治疗产品Zolgensma。除此以外,目前国内外还有10个基因治疗产品已被批准上市。截至2019年10月20日,ClinicTrails.gov 网站的数据显示全球已登记基因治疗临床试验总数为4091项,其中美国2198项,中国279项。
基因治疗产品正被研究用于治疗包括癌症、遗传病和传染病在内的疾病。
基因治疗有体内与体外两种途径,基因治疗产品按技术特点包括以下五种:
-
质粒DNA:环状DNA分子可以通过基因工程将治疗基因携带到人类细胞中。
-
病毒载体:病毒具有将遗传物质传递到细胞中的自然能力,因此一些基因治疗产品是从病毒中衍生出来的。一旦病毒被改造以消除它们引起传染病的能力,这些经过修饰的病毒就可以被用作载体(载体),将治疗基因携带到人类细胞中。
-
细菌载体:细菌可以被修饰以防止它们引起传染病,然后作为载体(载体)将治疗基因携带到人体组织中。
-
人类基因编辑技术:基因编辑的目的是破坏有害基因或修复突变基因。
-
患者源性细胞基因治疗产品:细胞从病人身上移除,基因改造(通常使用病毒载体),然后返回给病人。
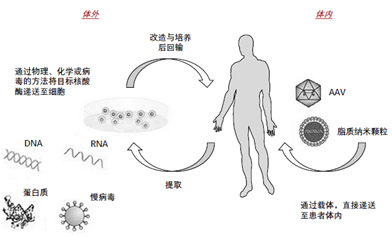
图1 基因治疗产品途径
随着对产品认识的深入和检测技术的发展,产品的质量研究应不断补充和完善并贯穿于整个生命周期。研究应采用先进、成熟的分析方法,全面了解产品质量属性,评估质量属性与产品安全性、有效性的相关性。
质量特性的研究应选用代表性工艺批次和适当生产阶段的样品作为研究对象,代表性批次如非临床研究批次、中试批次、关键性临床批次或商业化工艺批次等,生产阶段样品如原材料、中间体、原液和成品。原液和制剂之间若存在质量特性的差异,应分别取样进行分析。对于复核核酸载体,应对核酸载体、复核组分和最终复合物分别进行研究。
质量研究内容应覆盖所有可能与产品安全性、有效性相关的特性,一般包括结构、鉴别、一般理化特性、纯度、生物学活性、基因转导效率、杂质、基因型、表型等,具体研究项目应根据产品类型、作用机制、原材料和生产工艺决定。
结构的研究包括一级结构和高级结构。采用多种方法,如测序、限制性酶切谱图等,对载体完整的基因序列进行确认,尤其是目的基因以及相关的选择/调节/控制元件,基因中应不含有致癌或促瘤基因。
生物学活性的研究和方法学建立应依据产品适应症、给药途径和作用机制进行开发,尽可能建立与作用机制相同或相似的体、内外分析方法用于活性研究和质量控制。研究应能反应产品的转导活性,以及替代、补偿、阻断、修正特定基因的预期作用。含有多种活性成分的制品,需要分别建立方法对各个成分的活性进行测定,同时还应考虑活性成分之间可能存在的干扰或者协同作用。
产品相关杂质包括所有非目标或非功能形式的生产产物。病毒类载体一般应分析产物中的空壳病毒、错误包装病毒、杂合病毒、无活性病毒颗粒,病毒聚集体等杂质在产品中的残留水平,并对其安全性进行评估;核酸类载体一般应分析错误序列、不完整序列、降解片段、差异结构、错误修饰、或复杂递送系统错误组装组分等的残留水平;微生物载体一般应对株菌的单克隆性、质粒或改造基因丢失率等情况进行检定。
建立基因治疗产品物理数量和生物数量的检测指标,如:颗粒数、感染性滴度或感染性颗粒数、基因组DNA/RNA或质粒DNA浓度、细菌数等。病毒载体应对总颗粒数与感染性滴度或感染性颗粒数的比例进行控制,病毒载体类制剂建议以物理滴度计算临床计量。活的微生物载体应对活率,以及活菌与死菌的比例进行控制。分析应尽可能使用标准品或对照品来校准含量测定结果。
一般理化特定分析,如外观、澄清度、可见异物、不溶性微粒、pH值、渗透压等。此外,还可能要对病毒载体的复制能力、插入位点、质粒载体的转导率等。
质量标准的制定以控制最终产品的质量和批间一致性为目的,具体应根据工艺和控制需要对不同阶段的样品制定质量标准,一般包括原液,半成品(如有)和制剂的质量标准。质量标准的确定应基于质量研究,根据产品质量属性与安全性、有效性的相关性确定质量标准的具体内容,一般包括检验项目、分析方法和可接受标准。
标准限度一般应基于目标产品质量的设定、代表性工艺批次分析数据的统计、稳定性研究结果,以及人体或动物安全性研究数据等多个方面设定。
原液的质量标准一般包括外观、鉴别、理化特性、纯度、含量、活性、外源因子、内毒素、杂质(产品相关和工艺相关杂质),以及其他药典规定检测项目。
对于非复制型或条件复制型病毒载体,应对可复制型病毒进行检测和控制。除上述原液检测项目外,制剂质量标准还应关注受制剂处方、制剂生产工艺和包装容器影响的其他质量属性,一般还包括(但不限于):转导效率、感染活性、制剂外观、装量、可抽取体积、水分残留、制剂理化特性(如pH值、折光率、渗透压、不溶性微粒、可见异物等)、关键辅料含量(铝、复合辅料等)、制剂工艺杂质、可复制型病毒(如适宜),以及药典要求的制剂检项等。
制剂若采用特殊容器或药械组合装置,还需要根据装置的功能增加特定的放行检测。对于部分未纳入质量标准的检测项目,应说明其合理性,并提供验证依据。
基因治疗产品稳定性研究可参照《生物制品稳定性研究技术指导原则(试行)》和ICH Q5C的一般原则和相关要求进行,同时根据产品自身特点、临床用药情况等合理设计研究方案。
研究项目一般包括长期稳定性、加速稳定性、影响因素研究、运输稳定性、使用稳定性等,研究条件应根据具体保存、运输和使用情况,以及相应研究条件下研究结果的指示意义具体确定。研究应选用代表性工艺样品,装于代表性包装容器或使用器材进行。
稳定性研究监测项目应全面,尤其是对产品安全性、有效性、稳定性有重要指示意义的检测项目。研究期限应覆盖实际保存或使用时限。
容器和密闭系统一般包括原液、半成品(如有)、制剂的包装容器,生产过程中原材料、中间品的保存容器,以及生产过程中与产品直接接触的生产管线,如生物反应袋、一次性管线等。
为避免储存容器或密闭系统对产品的质量产生非预期影响,应对容器和密闭系统进行相容性研究、密闭性研究和安全性评估,特定条件下的密闭保存容器还应进行相应条件下的适用性研究,如冷冻适应性等。
对于具有特殊功能的次级包装材料,应将次级包装材料纳入容器和密闭系统的研究中,对其相应功能进行研究和验证。如涉及特殊给药装置,如电穿孔装置、鼻喷装置、无针注射器等,需提交相关研究资料或其他适用的支持资料。
参考文献:
[1] 基因治疗产品药学研究与评价技术指导原则(征求意见稿)
[2] 2021年中国基因治疗行业概览
本文为授权转载作品,仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
文中图片为授权正版图片,或来自微信公共图片库,或取自网络
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权






 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 