-
Beck A , Reichert J M. Antibody-drug conjugates[J]. mAbs, 2014, 6(1):15-17.
-
Diamantis N, Banerji U. Antibody-drug conjugates–an emerging classof cancer treatment[J]. Br J Cancer, 2016, 114(4): 362-367.
-
Bross P F, Beitz J, Chen G, et al. Approval summary: gemtuzumabozogamicin in relapsed acute myeloid leukemia[J]. Clin Cancer Res,2001, 7(6): 1490-1496
-
Bradley A M, Devine M, Deremer D. Brentuximab vedotin: an anti-CD30 antibody-drug conjugate[J]. Am J Health Syst Pharm, 2013,70(7): 589-597.
-
Adem Y T, Schwarz K A, Duenas E, et al. Auristatin antibodydrug conjugate physical instability and the role of drug payload[J].Bioconjug Chem, 2014, 25(4): 656-664.
-
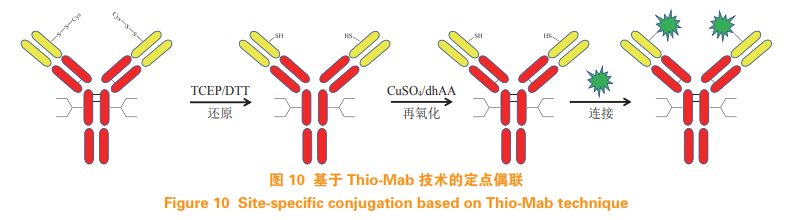
Junutula J R, Raab H, Clark S, et al. Site-specific conjugation of acytotoxic drug to an antibody improves the therapeutic index[J]. NatBiotechnol, 2008, 26(8): 925-932.
-
Wang L, Brock A, Herberich B, et al. Expanding the genetic code ofEscherichia coli[J]. Science, 2001, 292(5516): 498-500.
-
Hallam T J, Smider V V. Unnatural amino acids in novel antibodyconjugates[J]. Fut Med Chem, 2014, 6(11): 1309-1324.
-
Hallam T J, Wold E, Wahl A, et al. Antibody conjugates with unnaturalamino acids[J]. Mol Pharmaceutics, 2015, 12(6): 1848-1862.
-
Adumeau P, Sharma S K, Brent C, et al. Site-specifically labeledimmunoconjugates for molecular imaging—part 2: peptide tags andunnatural amino acids[J]. Mol Imaging Biol, 2016, 18(2): 153-165
-
Hutchins B M, Kazane S A, Staflin K, et al. Site-specific coupling andsterically controlled formation of multimeric antibody Fab fragmentswith unnatural amino acids[J]. J Mol Biol, 2011, 406(4): 595-603.
-
Axup J Y, Bajjuri K M, Ritland M, et al. Synthesis of site-specificantibody-drug conjugates using unnatural amino acids[J]. Proc NatlAcad Sci, 2012, 109(40): 16101-16106.
-
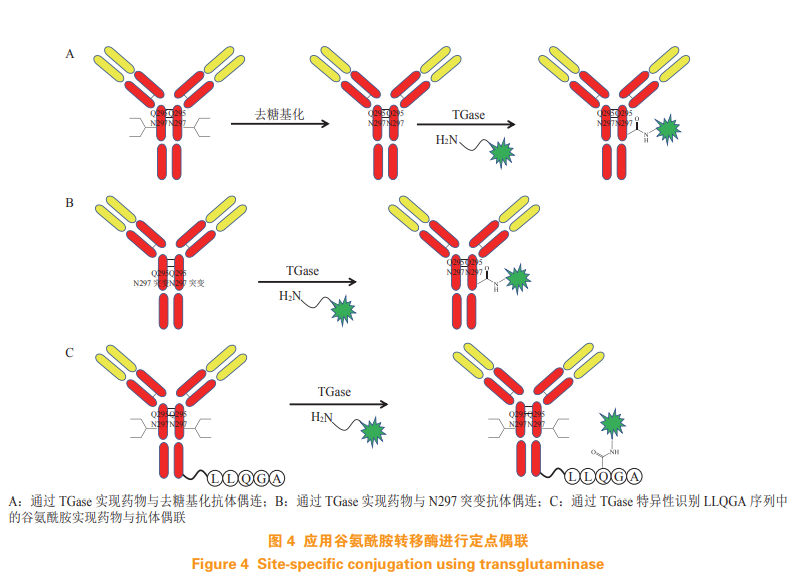
Jeger S, Zimmermann K, Blanc A, et al. Site-specific andstoichiometric modification of antibodies by bacterialtransglutaminase[J]. Angew Chem, 2010, 49(51): 9995-9997.
-
Dennler P, Chiotellis A, Fischer E, et al. Transglutaminase-basedchemo-enzymatic conjugation approach yields homogeneousantibody–drug conjugates[J]. Bioconjug Chem, 2014, 25(3): 569-578.
-
Strop P, Delaria K, Foletti D, et al. Site-specific conjugation improvestherapeutic index of antibody drug conjugates with high drugloading[J]. Nat Biotechnol, 2015, 33(7): 694-696.
-
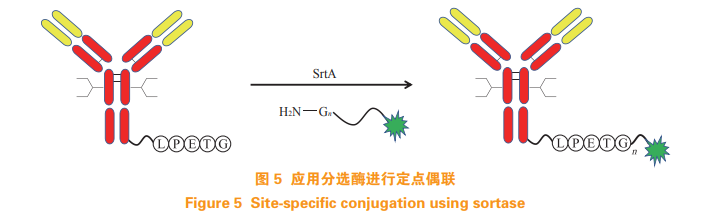
Beerli R R, Hell T, Merkel A S, et al. Sortase enzyme-mediatedgeneration of site-specifically conjugated antibody drug conjugateswith high in vitro and in vivo potency[J]. PLoS One, 2015, 10(7):e0131177. Doi: 10.1371/journal.pone.0131177.
-
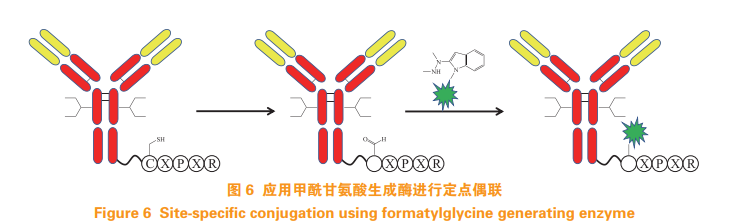
Drake P M, Albers A E, Baker J, et al. Aldehyde tag coupled withHIPS chemistry enables the production of ADCs conjugated sitespecifically to different antibody regions with distinct in vivo efficacyand PK outcomes[J]. Bioconjug Chem, 2014, 25(7): 1331-1341.
-
Rabuka D. Abstract 2662: site specific ADC generation usingSMARTag technology with programmable payload placement[J].Cancer Res, 2014, 74(Suppl 19): 2662. Doi: 10.1158/1538-7445.AM2014-2662.
-
Lee H W, Jeong Y H, Hwang J A, et al. Radioisotope-ADME studiesof Trastuzumab-monomethyl auristatin F in tumor bearing mice andhealthy marmosets[J]. J Biomed Res, 2020, 21(2): 79-90.
-
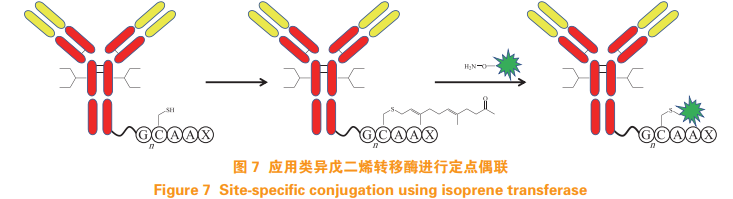
Van Berkel S S, Van Delft F L. Enzymatic strategies for (near) clinicaldevelopment of antibody-drug conjugates[J]. Drug Discov Today Technol, 2018, 30: 3-10.
-
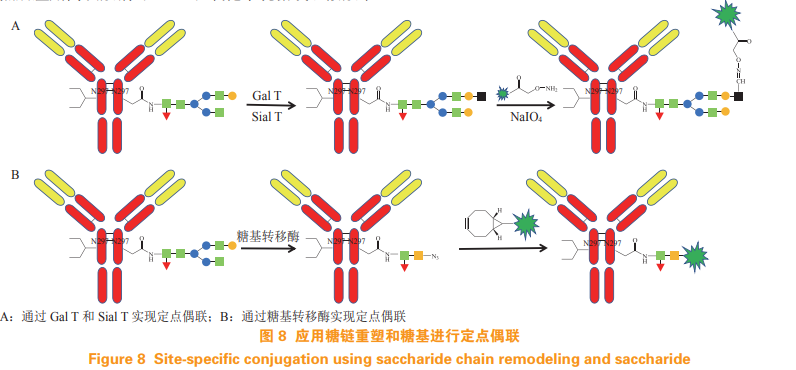
Zhou Q, Stefano J E, Manning C, et al. Site-specific antibody-drugconjugation through glycoengineering[J]. Bioconjug Chem, 2014,25(3): 510-520.
-
Zuberbuhler K, Casi G, Bernardes G L, et al. Fucose-specificconjugation of hydrazide derivatives to a vascular-targetingmonoclonal antibody in IgG format[J]. Chem Commun, 2012, 48(56):7100-7102.
-
Preparation of well-defined antibody-drug conjugates through glycanremodeling and strain-promoted azide-alkyne cycloadditions[J].Angew Chem Int Ed, 2014, 53(28): 7179-7182.
-
Geel R V, Wijdeven M A, Heesbeen R, et al. Chemoenzymaticconjugation of toxic payloads to the globally conserved N-glycan ofnative mabs provides homogeneous and highly efficacious antibodydrug conjugates[J]. Bioconjug Chem, 2015, 26(11): 2233-2242.
-
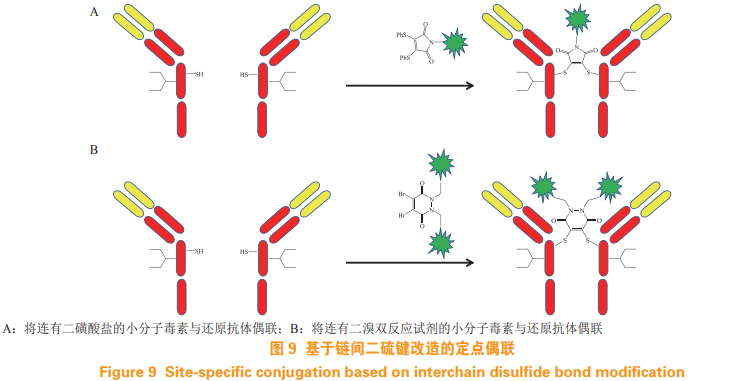
Morais M, Forte N, Chudasama V, et al. Application of next-generationmaleimides (NGMs) to site-selective antibody conjugation[J]. MethodsMol Biol, 2019, 2033: 15-24.
-
Schumacher F F, Nunes J P M, Maruani A, et al. Next generationmaleimides enable the controlled assembly of antibody-drugconjugates via native disulfide bond bridging[J]. Org Biomol Chem,2014, 12(37): 7261-7269.
-
Junutula J R, Raab H, Clark S, et al. Site-specific conjugation of acytotoxic drug to an antibody improves the therapeutic index[J]. NatBiotechnol, 2008, 26(8): 925-932.
-
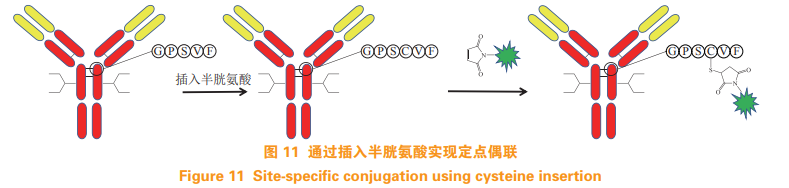
Dimasi N, Fleming R, Zhong H, et al. Efficient preparation of sitespecific antibody-drug conjugates using cysteine insertion[J]. MolPharm, 2017, 14(5): 1501-1516.
-
Shiraishi Y, Muramoto T, Nagatomo K, et al. Identification of highlyreactive cysteine residues at less exposed positions in the Fab constantregion for site-specific conjugation[J]. Bioconjug Chem, 2015, 26(6):1032-1040.
-
Shinmi D, Taguchi E, Iwano J, et al. One-step conjugation method for sitespecific antibody-drug conjugates through reactive cysteine-engineeredantibodies[J]. Bioconjugate Chem, 2016, 27(5): 1324-1331.
-
Li X, Patterso J T, Sarkar M, et al. Site-specific dual antibodyconjugation via engineered cysteine and selenocysteine residues[J].Bioconjug Chem, 2015, 26(11): 2243-2248.
-
McDonagh C F, Turcott E, Westendorf L, et al. Engineered antibodydrug conjugates with defined sites and stoichiometries of drugattachment[J]. Protein Eng Des Sel, 2006, 19(7): 299-307.






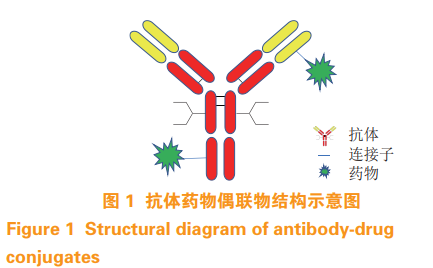
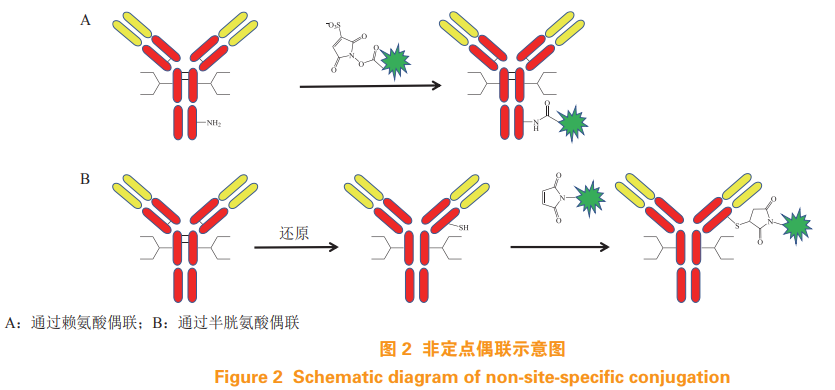
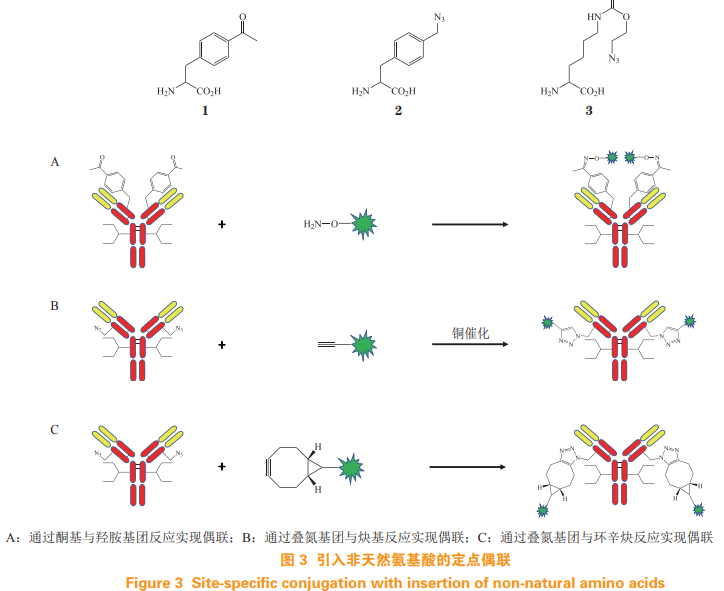






 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 