





中美新药定义的差异
美国新药定义
中国新药定义

中美新药审评的差异
审评程序和申报机制的差异
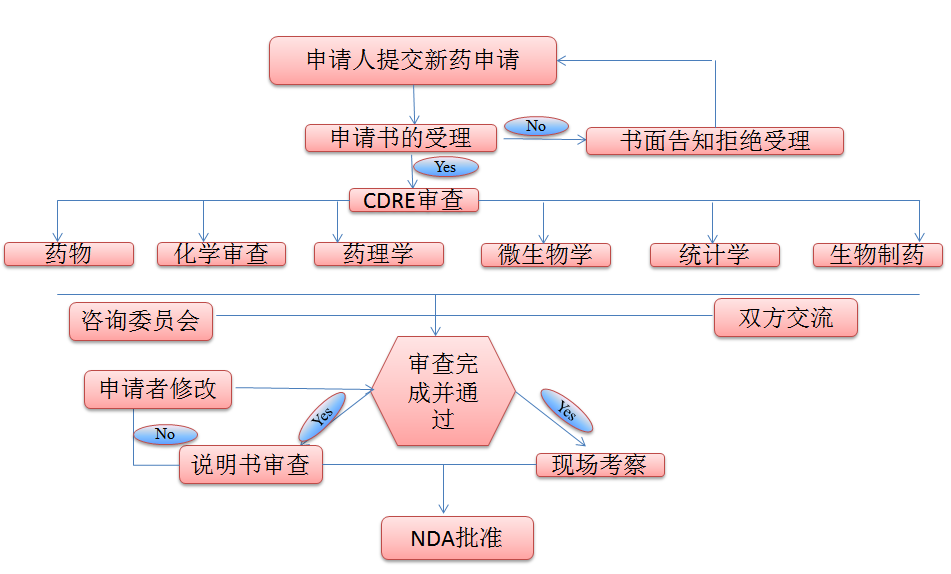
美国新药申请
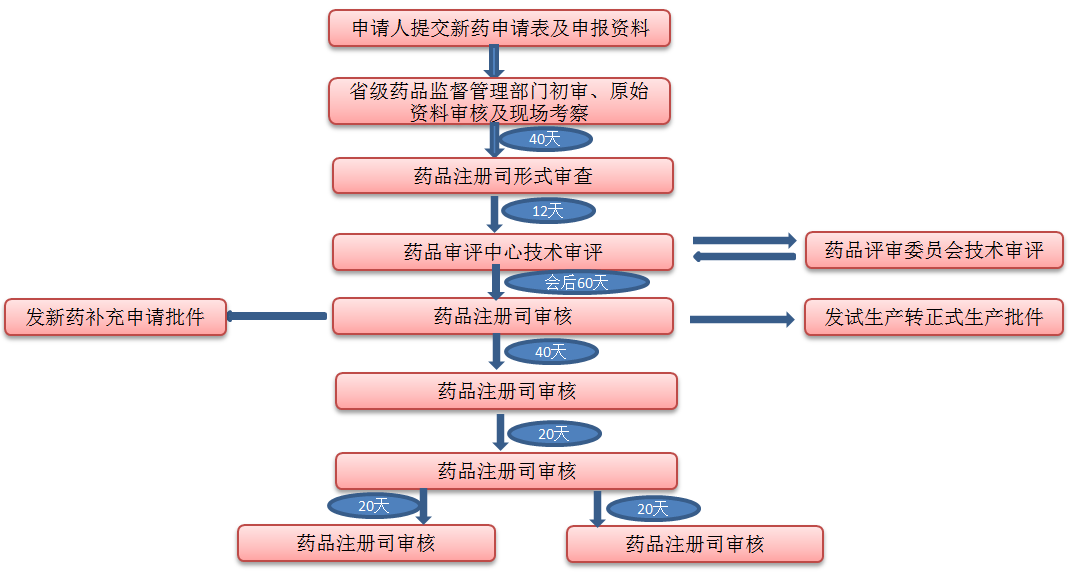
中国新药申请
审评原则的差异
再评价机制的差异
中美新药研究申请程序异同
相同点:
差异:
中美新药非临床研究申请异同
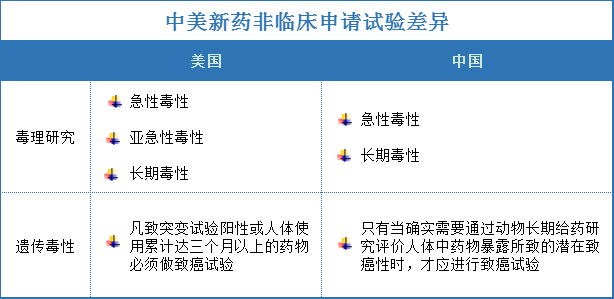
中美新药临床研究申请异同
相同点:


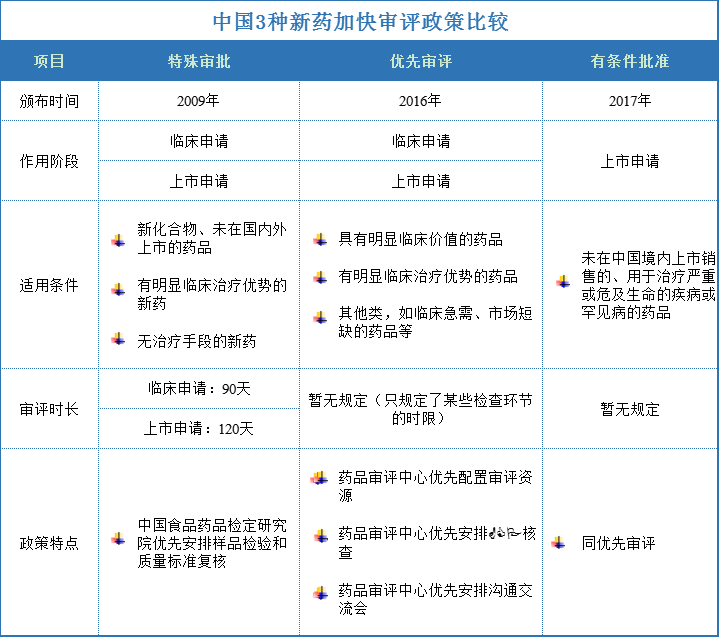
中美新药优惠政策的差异
美国市场独占权:

中国:
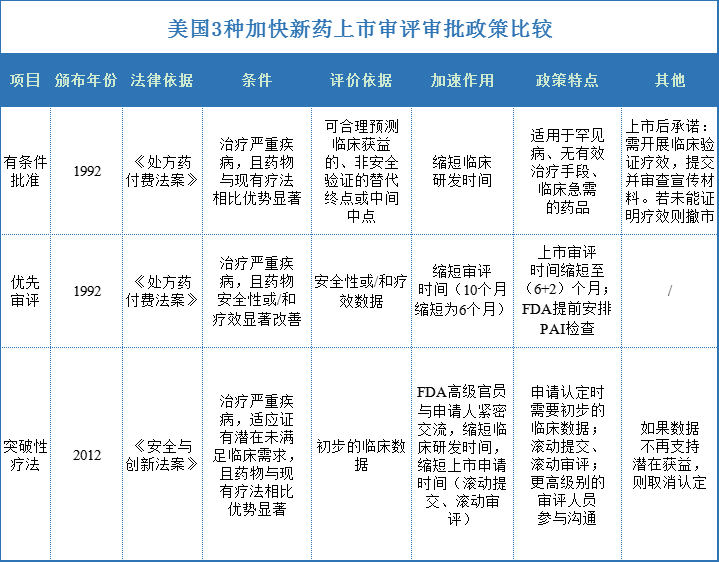
总结
参考文献:
[1]https://www.ecfr.gov/cgi-bin/text-idx?SID=a5e13772d51b5900dec2f52bb91c32e9&mc=true&node=se21.5.310_13&rgn=div8
[2]https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/fgwj/bmgzh/20070710010101571.html
[3]https://www.nmpa.gov.cn/directory/web/nmpa/zhuanti/lshzht/fzhypj/fzhyzhcfg/20150818101201803.html
[4] https://www.fda.gov/medical-devices/premarket-approval-pma/pma-review-process
[5]余菁菁. 中美新药审批制度的比较研究:一个经济学的视角.
[6]张琪, 颜建周, 马旭峰, 等. 美国药品上市后再评价法律制度实施的研究及其对我国的启示[J]. 中国药房, 2019, 30(15): 2017-2022.
[7]《药物警戒质量管理规范(征求意见稿)》.
[8]https://www.ecfr.gov/cgi-bin/retrieveECFR?gp=&SID=9d8edd80fb9c9d2e7149351a66421212&mc=true&n=sp21.5.312.b&r=SUBPART&ty=HTML#se21.5.312_122
[9] https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20100401145801553.html
[10]任晓星, 史录文. 中美欧新药上市加快审评审批政策研究[J]. 中国新药杂志, 2020, 29(9): 9621-971.



推荐阅读


 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 