
北京时间2025年8月20日,国际旗舰学刊《药理学与毒理学年鉴》(Annual Review of Pharmacology and Toxicology)以“Review in Advance”形式在线发表了由王明伟研究团队应邀撰写的题为“Structural Insights into the Development of Inhibitors against Cancer-Specific Mutations of PI3Kα”的长篇综述(图1,第66卷,2026年1月在美国正式出版)。该文系统梳理了PI3Kα致癌突变体(H1047R、E542K和E545K等)驱动的PI3K/AKT/mTOR信号通路异常激活机制,深度解析了从传统ATP竞争性抑制剂到新一代致癌突变体选择性变构抑制剂、共价抑制剂和蛋白降解剂的发展历程。文章特别阐述了致癌突变重塑PI3Kα三维结构及其动态特征,基此提出了设计高突变体选择性变构抑制剂的全新方案,旨在克服现有药物的毒副作用及耐药性。此篇力作不仅揭示了变构抑制剂的广阔应用前景,更为研发靶向PI3Kα的抗癌药物提供了新的思路。


图1. 论文首页标题
磷脂酰肌醇3-激酶(Phosphoinositide 3-kinase, PI3K)是一类催化磷酸肌醇磷酸化的脂质激酶,主要分为I、II和III三类1,2。其中,I类PI3K在响应细胞表面受体刺激的信号转导中发挥核心作用,并与癌症的发生发展密切相关;而II类和III类PI3K则主要参与膜转运过程3。哺乳动物表达四种I类催化亚型:p110α、p110β、p110δ(IA类)和p110γ(IB类)。它们通常与调节亚基形成异二聚体,催化生成关键第二信使PtdIns-3,4,5-P3(PIP3)。PI3K-AKT-mTOR 通路调控葡萄糖稳态、蛋白合成、细胞增殖与存活等关键生物学过程,是人类恶性肿瘤中最常见的异常信号通路之一4。其中编码p110α的基因PIK3CA的体细胞突变在多种癌症中普遍存在,超过80%的PIK3CA突变发生在三个特定位点:位于螺旋结构域的E542K 和E545K,以及位于激酶结构域的H1047R(图2a),使其成为重要的抗癌药物靶点。PI3K 抑制剂的研究始于20世纪90年代(图2b),早期抑制剂主要包括PI3K/mTOR 双重抑制剂和泛PI3K 抑制剂,但常伴随剂量毒性和脱靶效应,后期研发重点转向开发亚型选择性抑制剂和突变体选择性抑制剂(图2c)。目前,美国食品药品监督管理局(Food and Drug Administration, FDA)已批准6种 I类PI3K 抑制剂之临床应用,尤其在乳腺癌和血液系统恶性肿瘤(如淋巴瘤和白血病)的治疗中展现出显著的疗效。
该综述首先深入探讨I类PI3K在癌症信号转导中的作用,系统梳理了在其基础与临床研究中的里程碑事件(图2a)。随后,文章概述了小分子抑制剂的分类演进历程及其代表性药物(图2b)。在此背景下,结构生物学研究揭示了正构抑制剂因靶向保守ATP结合口袋具有局限性,由此凸显并推动了针对特定致癌突变体开发选择性抑制剂的迫切需求(图2c)。针对已报道的变构抑制剂,作者从结构生物学角度详细阐述了PI3Kα变构位点的多样性(图3),即便变构抑制剂的开发仍面临诸多挑战。最后,文章展望了包括PROTAC在内的新兴技术在克服毒性和耐药性方面的应用前景。总之,未来PI3K小分子抑制剂的发展应聚焦于减少毒副作用、提升致癌突变体选择性并改善治疗指数,进而达到更为安全有效的治疗目标。
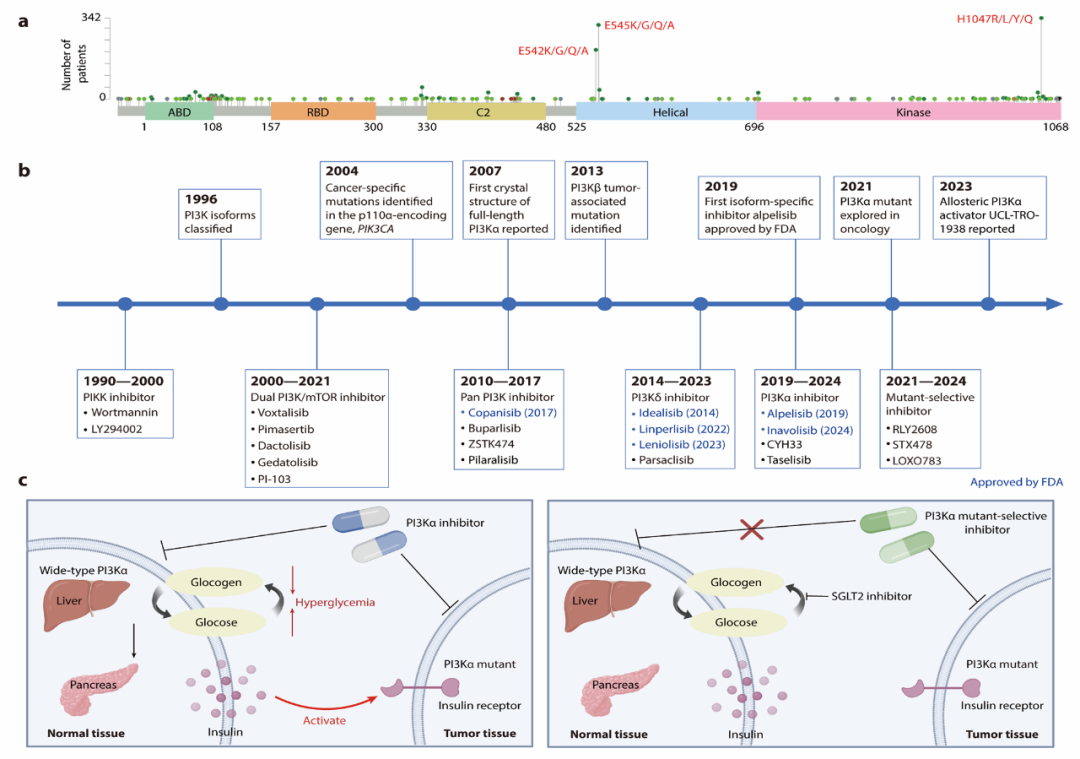
图2. Ⅰ类PI3K抑制剂的研究概况及发展历程。a.通过MSK-IMPACT分析阐明PI3KCA突变的频率分布,凸显E542、E545和H1047等位点突变频率最高。b. PI3K抑制剂开发之里程碑,FDA批准药物用蓝色表示。c.突变体选择性抑制剂特异性靶向PI3Kα突变的癌组织,从而避免非选择性抑制野生型PI3Kα引起的高血糖等副反应。
该文系统收集和整理了PI3Kα抑制剂的临床研究状况(补充资料表S2)。第一代PI3K抑制剂渥曼青霉素(Wortmannin)和LY294002因显著的毒性而只能用作工具药物5。后续研发的PI3K/mTOR双重抑制剂虽然在临床前模型中显示抗肿瘤活性,但其临床试验因严重不良事件或疗效有限而终止6。泛PI3K抑制剂Copanlisib于2017年获FDA批准用于复发滤泡性淋巴瘤,但依然存在高血糖等毒性反应7。鉴于四种亚型组织分布差异(p110α/β广泛表达,p110δ/γ分别在造血细胞和白细胞中高表达8),为了提高治疗指数、降低毒副作用,研发焦点进而转向了亚型选择性抑制剂。
Alpelisib 是一种高选择性PI3Kα抑制剂,于2019年获美国 FDA 批准与氟维司群联合治疗HR+/HER2–、PIK3CA突变的晚期或转移性乳腺癌成人患者,同时也可治疗因PI3K超级激活导致的过度生长综合征和血管畸形9。冷冻电镜结构解析显示,Alpelisib特异性作用于PI3Kα催化位点,从而阻断 ATP 和磷脂酰肌醇的结合10;在 E542K和H1047R 突变体中,其结合模式虽有细微调整但仍保持效力11。CYH33 是一种对PI3Kα具有高选择性的吡咯并苯并三嗪衍生物。Ia期临床试验显示,PIK3CA突变的晚期实体瘤患者之客观缓解率达14.3%,且毒性可控12。尽管亚型选择性抑制剂取得进展,其剂量限制性毒性(如高血糖和口腔黏膜炎的发生率超过50%)依然显著,这与PI3K/AKT/mTOR 通路在代谢调节和上皮细胞稳态中的关键角色密不可分。此外,ATP 结合口袋在野生型和突变体间的高度保守性导致治疗窗狭窄;耐药机制则常涉及 ATP 口袋的二次突变或代偿性信号通路激活13。这些瓶颈提示精准靶向突变体及超越 ATP竞争性抑制的必要性。
变构抑制剂代表了一种靶向致癌性PI3Kα突变的新策略。目前已报道的变构抑制剂包括RLY-2608、STX-478、QR-7909、QR-8557、Compound 17、LOXO-783、OKI-219和CGT4824等。通过对冷冻电镜、X射线晶体学和分子动力学模拟等技术获得的高分辨率结构进行比较分析(图3),该文明确揭示了正构与变构抑制剂作用模式之差别:正构抑制剂(如Alpelisib、Inavolisib和Cpd 19)均占据高度保守的ATP结合口袋(图3a)并与特定氨基酸残基作用,其选择性差异主要源于配体特异性相互作用而非ATP口袋构象改变。而变构抑制剂靶向ATP结合位点外的三个独特口袋:变构口袋1(QR-7909占据,图3b)位于激活环C末端,富集芳香残基,通过π–π堆积和疏水作用等实现其对H1047R的高选择性14;变构口袋2(RLY-2608和 STX-478等占据,图3c)邻近激活环,通过保守的极性相互作用及配体特异性相互作用将PI3Kα锁定在失活状态;变构口袋3(UCL-TRO-1938占据,图3d)通过极性作用等促进激活环构象重排15。这些结构研究显示变构抑制剂利用突变特异性构象和隐蔽位点获得高选择性和低毒副作用之优势,但是其后继开发仍然面临缺乏系统性发现新位点方法、难以捕获柔性突变构象和出现潜在脱靶效应等困局16。未来需整合计算生物学与高分辨结构解析技术,深入研究突变特异性变构机制,并结合功能验证来推进激酶相关疾病精准变构疗法的开发。
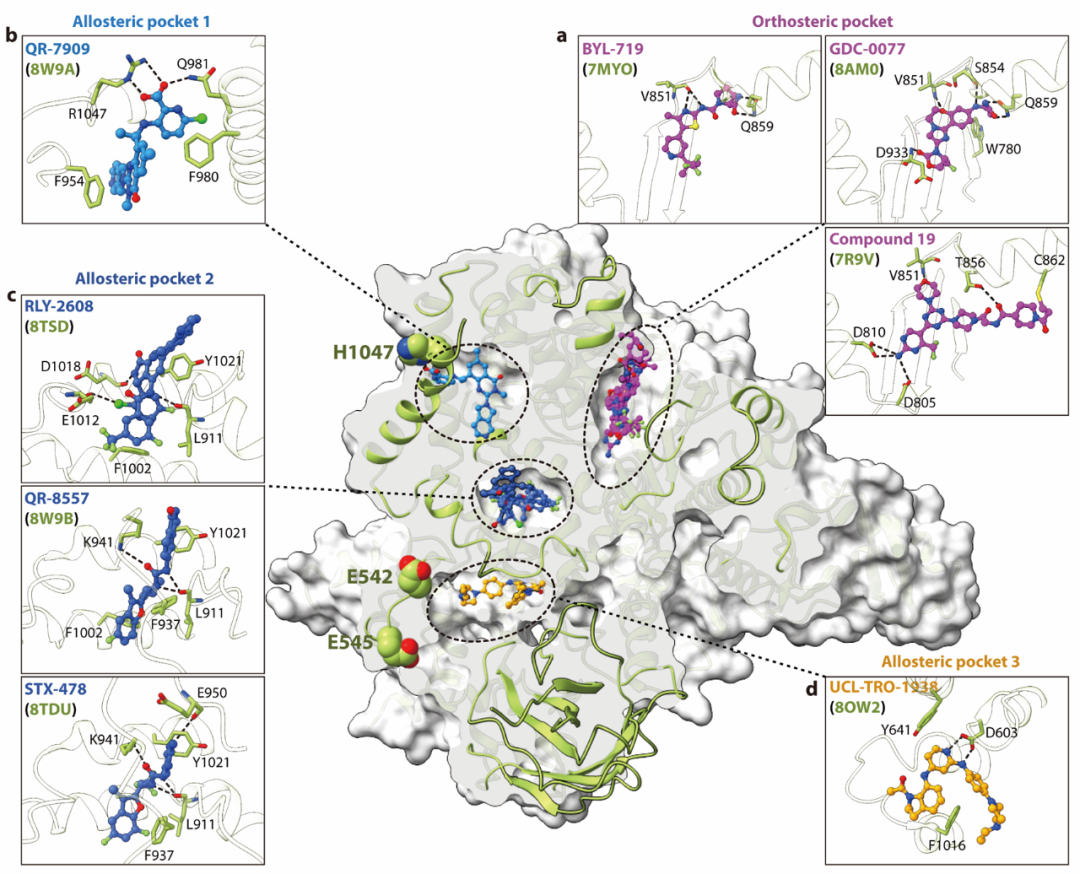
图3. PI3Kα的正构位点和变构位点对比图。a. ATP结合位点即正构配体结合口袋。b. 变构口袋1。c. 变构口袋2。d. 变构口袋3。
该综述最后展望了这一领域的前沿发展方向。一方面,当前研究正超越传统的正构/变构抑制模式,探索靶向蛋白降解、共价修饰和蛋白–蛋白相互作用等创新策略。其中,高效PI3Kα降解剂Inavolisib能够选择性降解突变蛋白,其III期临床试验在特定患者群体中疗效显著,已获得FDA突破性疗法的认定。另一方面,为了应对耐药性挑战,组合疗法(如与免疫检查点抑制剂或组蛋白去乙酰化酶抑制剂联用)以及基于PROTAC技术的新型双功能分子应运而生17,未来将在提高PROTAC分子的细胞渗透性和生物利用度上发力,不断优化临床治疗方案。
近年来,王明伟团队与美国Scripps研究所Peter K. Vogt教授紧密合作,先后解析了全长野生型PI3Kα和3个致癌突变体等的冷冻电镜结构,在《美国国家科学院院刊》(PNAS)上连发4篇研究论文,取得了开创性的突破10,11,18-20。此次是《药理学与毒理学年鉴》年初首发该团队有关减重药物研发进展综述(https://doi.org/10.1146/annurev-pharmtox-061324-011832;IF=13.1)后的又一篇重磅长文。
复旦大学基础医学院博士研究生刘晓为本文的第一作者;共同作者包括三亚深海化合物资源中心研究助理陈燕艳、上海交通大学医学院附属瑞金医院医药结构生物学转化研究中心博士后韩伟、陈彦、冯文博和副研究员周庆同;上海交通大学医学院附属瑞金医院医药结构生物学转化研究中心课题组长、三亚深海化合物资源中心主任、复旦大学基础医学院“复旦讲座教授”王明伟为通讯作者。这项工作获得了国家自然科学基金委员会、海南省科技重大专项、国家科学技术部和上海市科学技术委员会等机构的经费资助。
全文链接:
https://www.annualreviews.org/content/journals/10.1146/annurev-pharmtox-082924-022724
参考文献:
1 Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy.Cancer Sci 2008;99:1734-40.
2 Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease.Cell 2017;170:605-35.
3 Li H, Wen X, Ren Y, Fan Z, Zhang J, He G, et al. Targeting PI3K family with small-molecule inhibitors in cancer therapy: current clinical status and future directions.Mol Cancer 2024;23:164.
4 Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT.Curr Opin Cell Biol 2017;45:62-71.
5 Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling.Nat Rev Mol Cell Biol 2010;11:329-41.
6 Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, DeLuca J, et al. A Phase Ib Study of BEZ235, a Dual Inhibitor of Phosphatidylinositol 3-Kinase (PI3K) and Mammalian Target of Rapamycin (mTOR), in Patients With Advanced Renal Cell Carcinoma.Oncologist 2016;21:787-8.
7 Munoz J, Follows GA, Nastoupil LJ. Copanlisib for the Treatment of Malignant Lymphoma: Clinical Experience and Future Perspectives.Target Oncol 2021;16:295-308.
8 Zhao W, Qiu Y, Kong D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy.Acta Pharm Sin B 2017;7:27-37.
9 Tankova T, Senkus E, Beloyartseva M, Borstnar S, Catrinoiu D, Frolova M, et al. Management Strategies for Hyperglycemia Associated with the alpha-Selective PI3K Inhibitor Alpelisib for the Treatment of Breast Cancer.Cancers (Basel) 2022;14:1598.
10 Liu X, Yang S, Hart JR, Xu Y, Zou X, Zhang H, et al. Cryo-EM structures of PI3Kalpha reveal conformational changes during inhibition and activation.Proc Natl Acad Sci U S A 2021;118:e2109327118.
11 Liu X, Zhou Q, Hart JR, Xu Y, Yang S, Yang D, et al. Cryo-EM structures of cancer-specific helical and kinase domain mutations of PI3Kalpha.Proc Natl Acad Sci U S A 2022;119:e2215621119.
12 Wei XL, Liu FR, Liu JH, Zhao HY, Zhang Y, Wang ZQ, et al. First-in-human phase Ia study of the PI3Kalpha inhibitor CYH33 in patients with solid tumors.Nat Commun 2022;13:7012.
13 Varkaris A, Fece de la Cruz F, Martin EE, Norden BL, Chevalier N, Kehlmann AM, et al. Allosteric PI3Kalpha Inhibition Overcomes On-target Resistance to Orthosteric Inhibitors Mediated by Secondary PIK3CA Mutations.Cancer Discov 2024;14:227-39.
14 Huang X, Wang K, Han J, Chen X, Wang Z, Wu T, et al. Cryo-EM structures reveal two allosteric inhibition modes of PI3Kalpha(H1047R) involving a re-shaping of the activation loop.Structure2024;32:907-17 e7.
15 Gong GQ, Bilanges B, Allsop B, Masson GR, Roberton V, Askwith T, et al. A small-molecule PI3Kalpha activator for cardioprotection and neuroregeneration.Nature 2023;618:159-68.
16 Lu S, Shen Q, Zhang J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms.Acc Chem Res 2019;52:492-500.
17 Cirillo D, Diceglie M, Nazare M. Isoform-selective targeting of PI3K: time to consider new opportunities? Trends Pharmacol Sci 2023;44:601-21.
18 Hart JR, Liu X, Pan C, Liang A, Ueno L, Xu Y, et al. Nanobodies and chemical cross-links advance the structural and functional analysis of PI3Kalpha.Proc Natl Acad Sci U S A 2022;119:e2210769119.
19 Zhou Q, Liu X, Neri D, Li W, Favalli N, Bassi G, et al. Structural insights into the interaction of three Y-shaped ligands with PI3Kalpha.Proc Natl Acad Sci U S A 2023;120:e2304071120.
20 Krimmer SG, Schlessinger J. Unveiling molecular insights into the mechanism of activation of oncogenic phosphoinositide 3-kinase mutants.Proc Natl Acad Sci U S A 2022;119:e2218237119.
本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 