



辉瑞研究人员近日JCIM的文章 (之前已有预印本公开)。
评估小分子的构象是否低能,是药物设计很重要的一个方面,但有时 (也许是往往) 并没有很好被考虑。
docking打分或者没有考虑小分子构象penalty,或者有strain energy,但是基于MM方法,准确性不佳。
QM方法算得的结果比较准确,但计算量大,通量低,只适合精细modeling,不适合大规模虚拟筛选。
用机器学习/深度学习来“学习”训练集 (QM数据或晶体结构) 的信息并建立model,来预测新的小分子的低能构象,之前已有报道,这篇文章也是类似的尝试。
小分子torsion是影响小分子构象一个重要方面,这篇文章主要集中在小分子torsional energy profiling上面。
也就是CADD或基于结构的药物设计常说的扫二面角,比如前几天提的BI大环EGFRi那篇文章就有。现在比较多的用QM方法扫,也有用MM方法的。
当然还有一种方法是查晶体数据库,通过statistics方法看优势torsion的分布,如果数据库中有较多相似结构的话。
回到文章。
方法部分简单介绍 (图1, 图2),个人也不在行。
首先是为AI找学习素材。
辉瑞研究人员将内部分子库碎片化,拆成保留torsional特征的最小片段。
从中挑一部分最常见的片段,先用MM方法找出不同torsion angle的构象,然后对这些构象依次用QM方法 (B3LYP/6-31G**) 算对应的能量,这一步计算量大,放在亚马逊云上进行。
用这些数据来“喂”给AI,建模用到两种方法,随机森林及DNN (Deep Neural Network),期间也有迭代。模型建好后,进一步验证及预测。

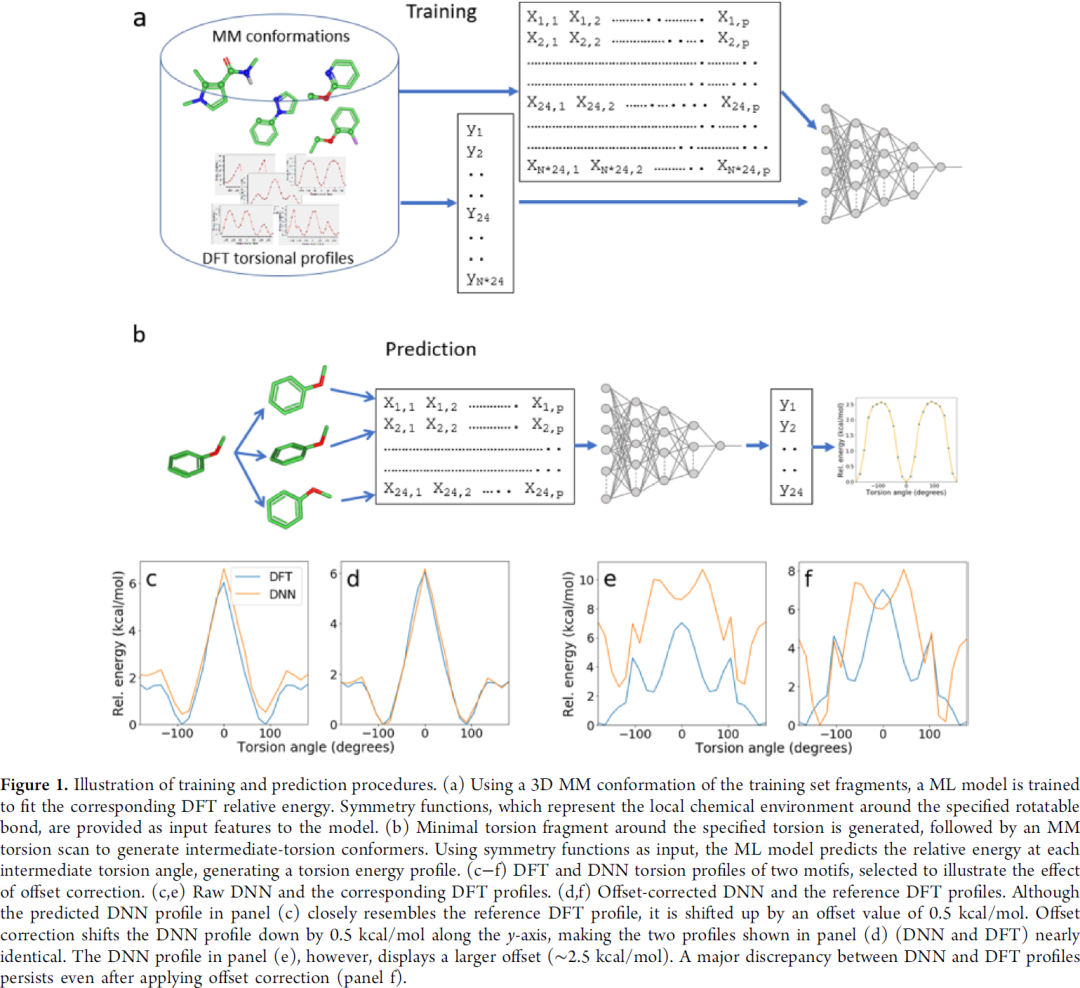
图2 c d e f,其中c e是校正前的结果,d f做了简单校正,即把橘线 (DNN) 整体下拉,让最低点贴近x轴。c的结果很不错,校正后与DFT结果基本重叠,E的结果就比较差,趋势都不对。
着重看结果部分。
整体来讲,与DFT结果还是有比较好的相关性,Pearson相关系数0.94,平均绝对误差0.7 Kcal/mol,RMSE 1.2 Kcal/mol。
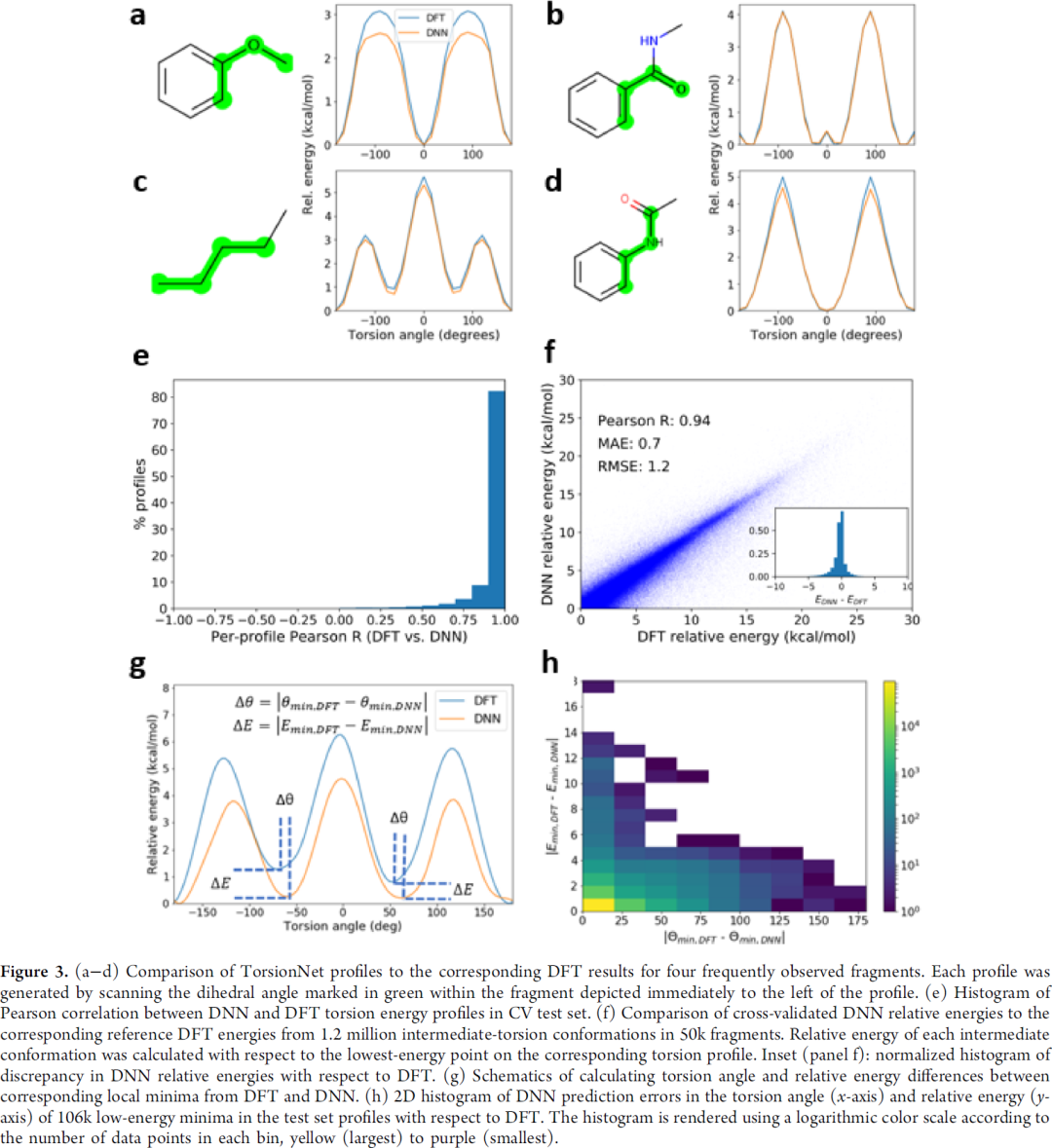
图4是与晶体结构的验证,曲线能量低的点,晶体结构分布多,整体来讲吻合也比较好。
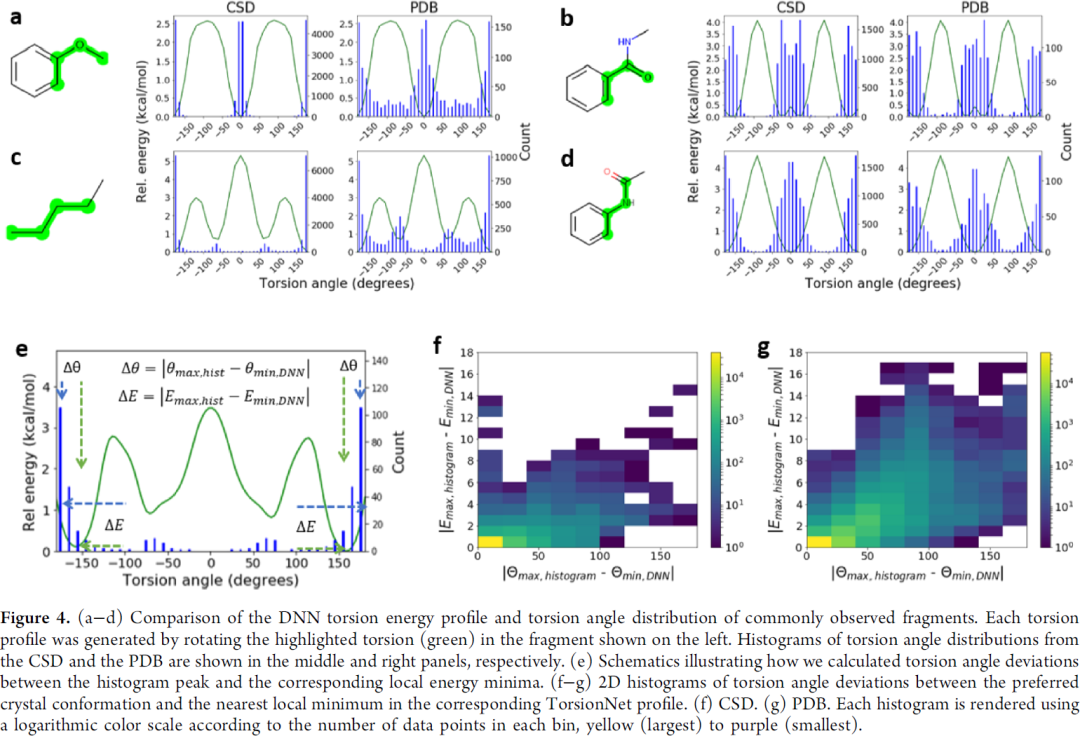
图5是统计学上探讨strain energy与分子活性的关系,当然分子活性不完全由strain energy决定,但可以看出分子活性与预测的strain energy有相关性,整体来讲活性差的分子strain energy更高。
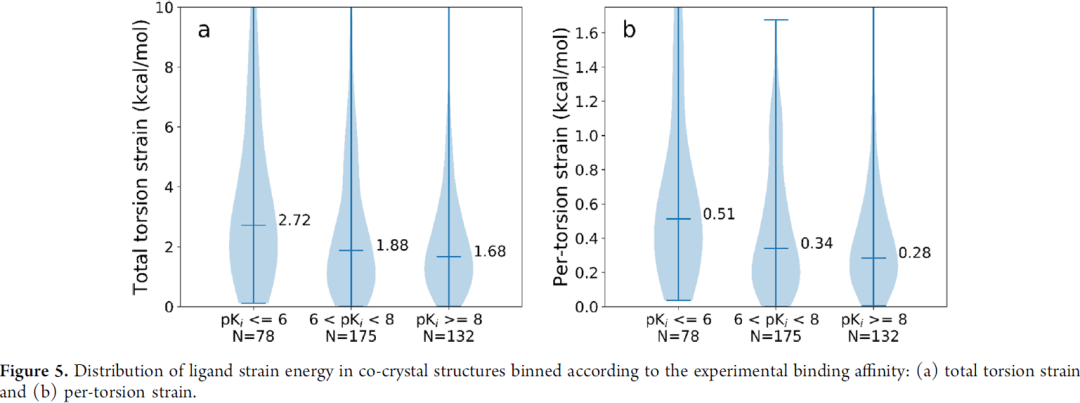
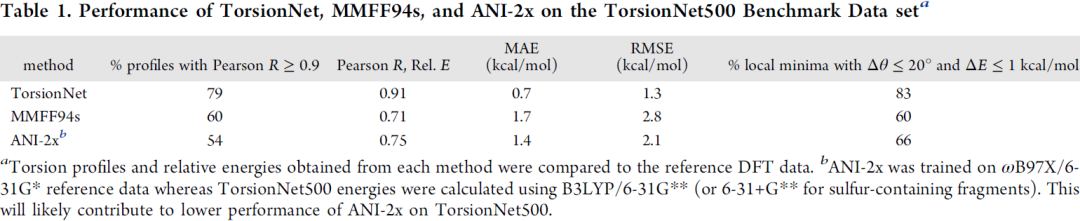
图6是三种方法在benchmark数据集的比较,TorsionNet即这篇文章的方法,MMFF94是较常见的MM力场,ANI-2x是之前发表的AI类力场方法,可以看出TorsionNet表现相对最好,ANI-2x有些指标反而不如MMFF94,作者认为可能是因为这个模型训练时用的QM方法不同。
最后是三个应用场景。
第一个是从一系列较低能构象中挑出晶体结构pose,这个也许在晶型预测有潜在应用,图7可以看出,文章model比MMFF力场表现要好一些,尤其在CSD数据集 (在PDB数据集表现相对差可以理解)。
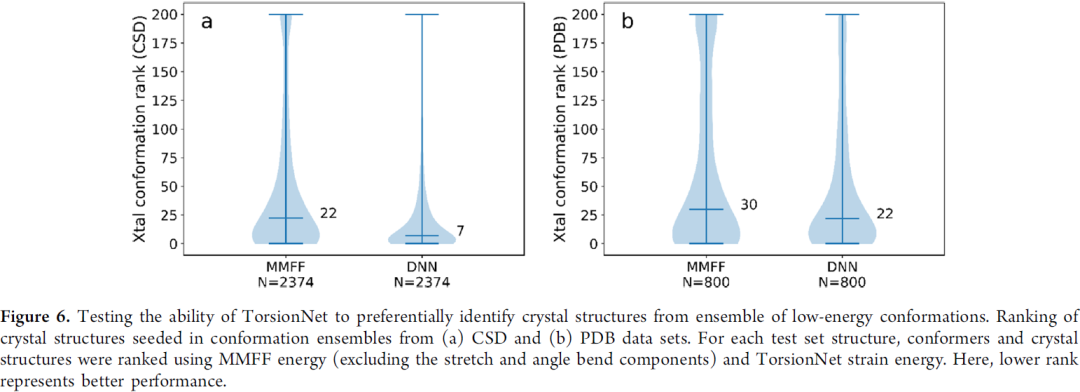
图7
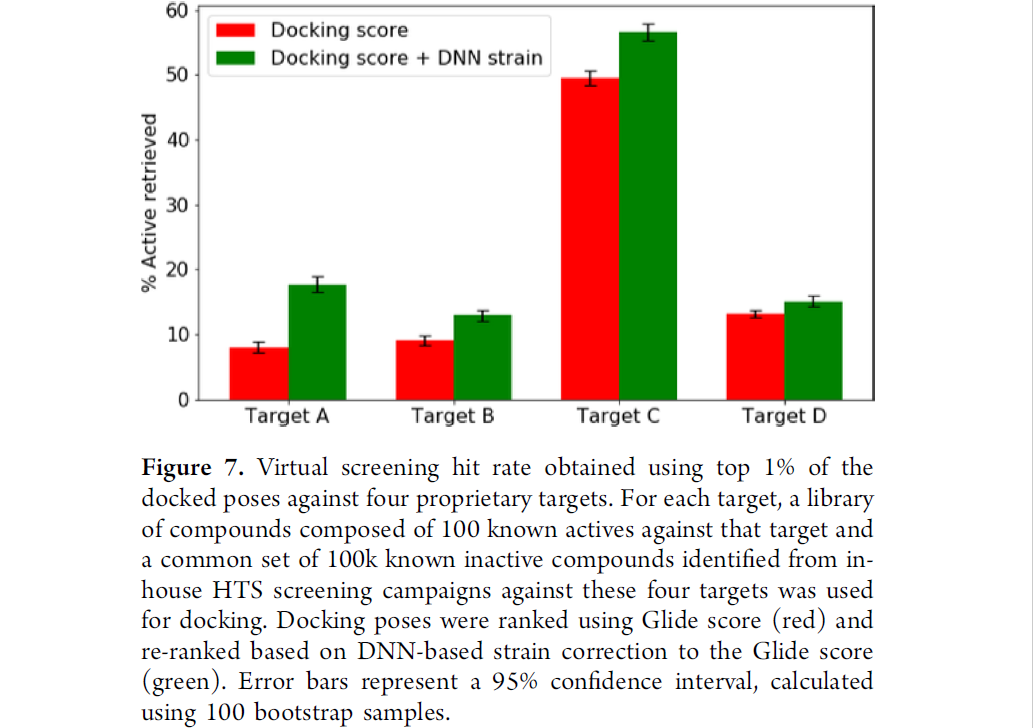
结果如图8所示,加上DNN strain校正的打分函数,有相对更高的虚筛命中率。另外注意这里dataset包含较多高活性分子,因此命中率普遍偏高,不能代表真实世界VS水平。
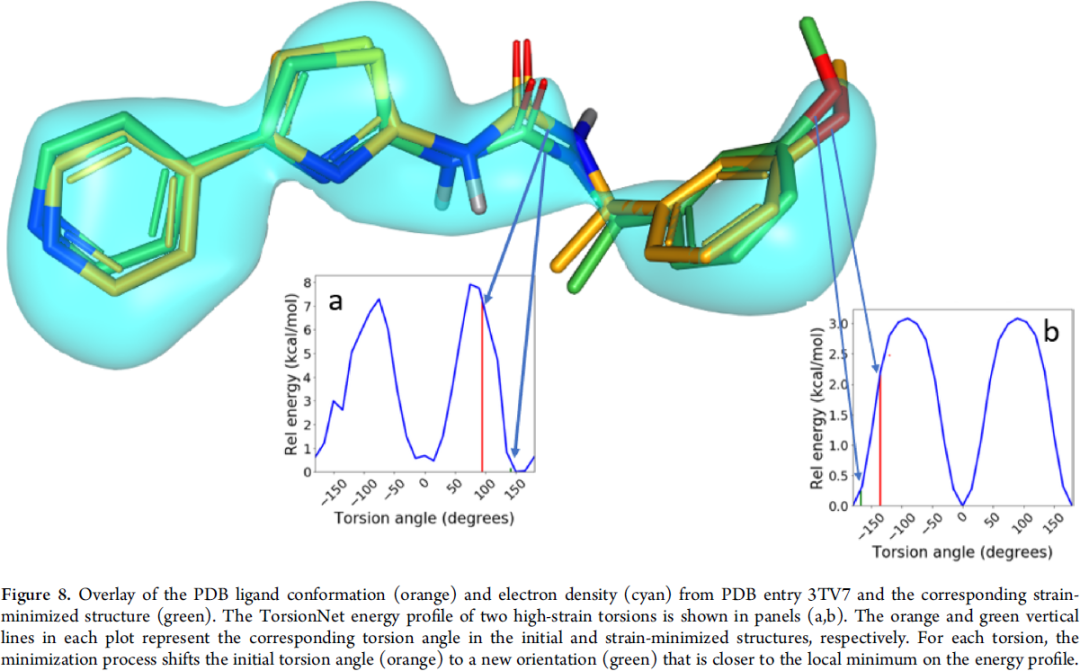
第三个是用来refine晶体结构小分子的构象,如图9所示,青色的部分表示晶体的density map,橘色的是原先fit的小分子pose (一般是根据MM力场),但有些地方看起来不是低能的,而较低能的构象也能fit进这个map,没有其它数据支持的话应该采纳这个更低能的构象 (refine后的绿色构象map缺失那块感觉也还不低能,可能需要进一步探讨)。
总之,这篇文章探讨了用deep learning方法预测小分子torsional energy,截取了小分子构象研究中的一部分话题来研究。结果显示,DNN model预测的结果比一般的MM力场方法要好,而比精度更高的QM方法要快,有可能用于改善docking score等应用。
相关code作者上传在https://github.com/PfizerRD/TorsionNet,不过相关的数据集 (5万个fragment及QM计算结果) 只公开了一部分 (500个) 作为benchmark。另外,MM conformation这一步用的是个商业软件,用其它软件应该也可代替,但有可能最终的performance会有不同。
TorsionNet: A Deep Neural Network to Rapidly Predict Small- Molecule Torsional Energy Profiles with the Accuracy of Quantum Mechanics. JCIM, Feb. 4, 2022.
End
版权声明/免责声明
本文为授权转载文章,仅代表作者观点,版权归作者。
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!

推荐阅读
回望创新药的2021:内卷之下,行业呼唤差异化创新 朗盛投资&药时代联合举办——思南峰会系列分享之“再论CD47” 里程碑ODAC会议召开之际,FDA Pazdur主任再次表态:不针对中国,要求临床研究必须能够代表美国 2022年新药产业链众生图 赛诺菲启用全新企业品牌和logo!附:辉瑞、百时美施贵宝、百济神州、和黄医药之宣传片 FDA大院里的华人 | 王亚宁博士和王亚平博士 震惊!虎年第一挖!辉瑞“突袭”罗氏总部,从CEO身边挖人,Pao博士将出任开发主管;Hans Clevers教授将加盟罗氏 Protocol撰写实操训练营!26节课,助您直达国际标准! 国际临床研究超级训练营!40节课,帮您系统学习国际临床试验前沿设计方法! 虎虎生威!6个视频,35篇文章,助您轻轻松松了解火热的Protac! FDA前后大变脸,中国创新药出海恐生难?

 点击这里,与~20万同药们喜相逢!
点击这里,与~20万同药们喜相逢!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 