




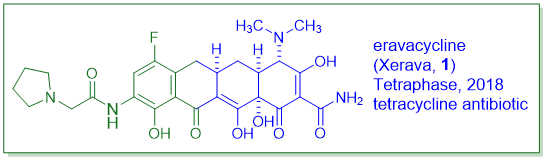
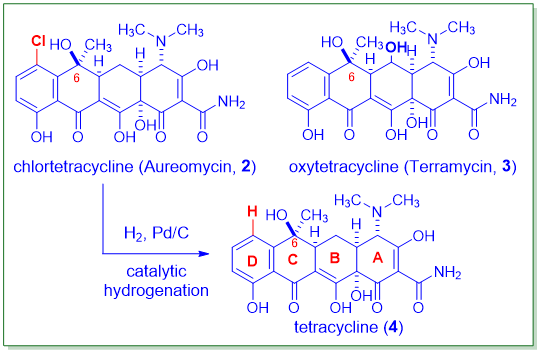
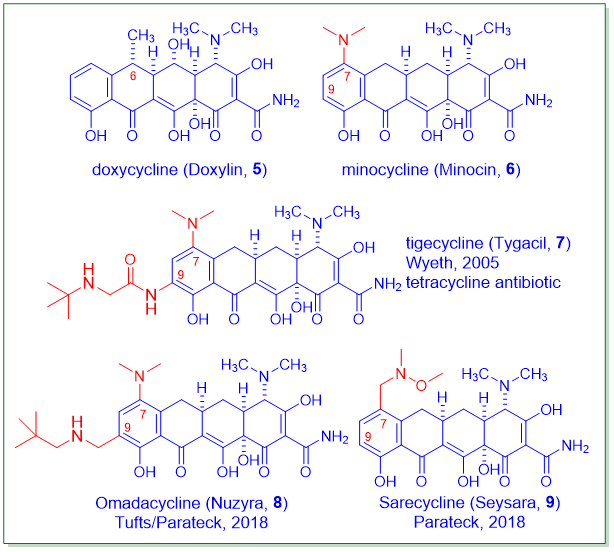
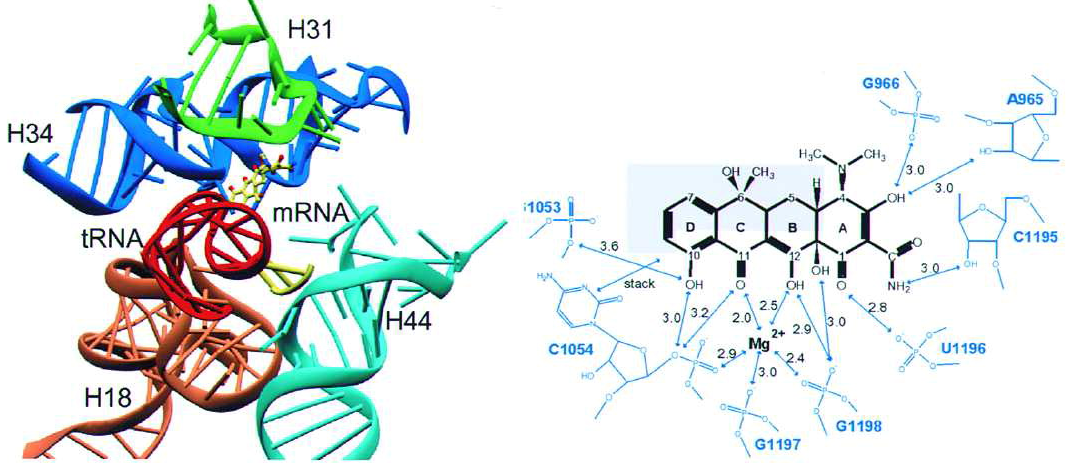
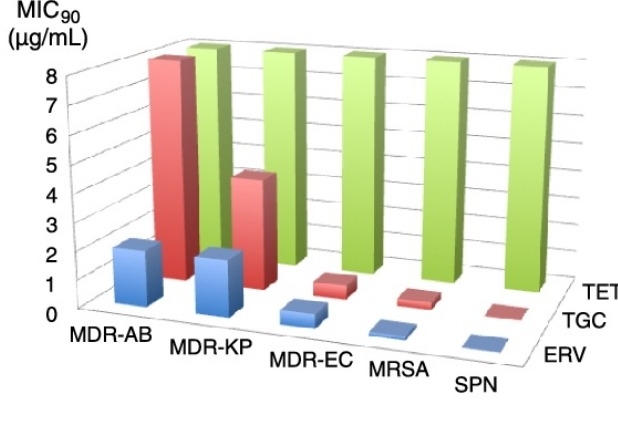
“四环素抗生素在20世纪40年代开始使用。近80年来,许多细菌病原体普遍对四环素产生耐药性。幸运的是,Tetraphase Pharmaceuticals研发的一种完全合成的四环素抗生素Eravacycline(Xerava,1)于2018年获得FDA批准。与现有的其他四环素相比,它具有超强的效力,是对抗细菌感染“武器库”中的一大补充。”
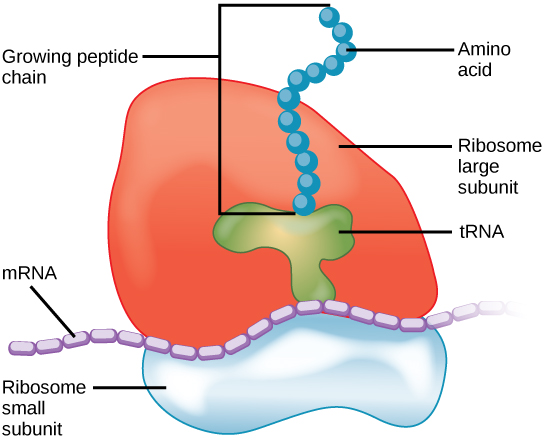
四环素作为抗生素如何起作用?



推荐阅读
应对新冠除了疫苗,还有药物。Cyclica运用人工智能技术发现“capmatinib”老药新用! 中国好BD|北海康成(CANbridge) 李文辉博士获颁巴鲁克•布隆伯格奖 祝贺!2021年中国国家药监局(NMPA)“官宣”批准的6款创新药 药时代倡议 | 每年5月21日成为“世界感谢患者日”(IPAD)! 灵魂拷问!谁“动了”印度民众的疫苗? PPT分享 | WHO:国药中生、科兴生物灭活疫苗临床数据评估报告 国内PARP抑制剂三足鼎立局面将被打破,百济神州帕米帕利获批在即 新冠疫苗开发被誉为诺曼底登陆:美国十个月上市两款核酸疫苗的奇迹是如何发生的? 疫苗大规模注射四个月后,美国的疫情怎么样了?(最新版本)阅读量已5.8万+ 药时代故事汇|Keytruda、勃林格殷格翰、礼来、复宏汉霖、亚盛。。。 10万+励志长文!坐了40年冷板凳,现在拯救美国就靠她!(附:百条精选点评) 收藏 | 拯救了美国的mRNA疫苗,制造过程首次揭秘 收藏 | 4月份FDA和NMPA批准新药汇总


 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 