


正文共: 5186字 3图
预计阅读时间: 13分钟
北京时间2023年8月8日,复旦大学基础医学院王明伟团队携手中国科学院上海药物研究所杨德华团队在《美国国家科学院院刊》(Proceedings of the National Academy of Sciences of the United States of America, PNAS)在线发表了题为“Structural analysis of the dual agonism at GLP-1R and GCGR”的研究成果(图1)。这篇由2012年诺贝尔化学奖得主Robert J. Lefkowitz教授主审的PNAS直投(Direct Submission)论文报道了胰高血糖素样肽-1受体(Glucagon-like peptide-1 receptor, GLP-1R)和胰高血糖素受体(Glucagon receptor, GCGR)双重激动剂激活受体的冷冻电镜结构,揭示了三个多肽发挥不同双重药理活性的分子机制,为设计和开发更为有效的多重激动剂提供了新的思路。

图1. 论文首页标题
GLP-1R和GCGR属于B1类G蛋白偶联受体(G protein-coupled receptor, GPCR),是维持人体血糖平衡的重要“调节器”。其中GLP-1R广泛分布于全身多个脏器,除胰腺外还包括中枢神经系统、胃肠道、心血管系统和脂肪组织等。因此,GLP-1R激动剂通过多种生理作用如促进胰岛素分泌、抑制胰高血糖素分泌、减低食欲、增加饱腹感和延缓胃排空等来达到降糖效果,是治疗2型糖尿病和肥胖症的明星靶标1。而GCGR主要在肝脏表达,其次是肾脏、心脏、脂肪组织、胰岛和中枢神经系统等。胰高血糖素在促进肝脏糖原分解、糖异生和脂质代谢的同时,对胰岛素分泌、食欲和氨基酸代谢也有一定的调控作用2。开发GLP-1R/GCGR双重激动剂的目的在于同时激活GLP-1R和GCGR进而产生叠加效应,为相关代谢性疾病的治疗提供新的手段。
预计至2035年,全球2型糖尿病患者将达5.9亿人,到2045年将跃升至7 亿人3,而肥胖症的发病率更是逐年增加,预计2030年欧美一些发达国家的超重人群将超过人口的30% 4。正是由于持续扩大的市场需求,凭借司美格鲁肽的减肥奇效,使得GLP-1R靶点火爆全球。近年来,除 GLP-1R 单靶点药物以外,全球各大医药公司也在角逐GLP-1R/GIPR/GCGR多靶点激动剂领域。其中,礼来的替西帕肽(Tirzepatide)在 GLP-1R/GIPR双重激动剂开发上一骑绝尘,具有显著的降糖减重效果,已获得 FDA 批准上市5。或许由于礼来在 GLP-1R/GIPR 领域的领先优势,其他药企的布局似乎更偏向于开发GLP-1R/GCGR双重激动剂:尽管目前已有多个候选药物进入临床试验,但由于激动GCGR对血糖造成的复杂影响阻碍了它们的开发进程。Peptide 15是第一个被报道的GLP-1R/GCGR强效双重激动剂,对GLP-1R和GCGR均有类似于内源性配体的激动作用,然而却因高血糖副作用而未能进入临床研究阶段6;赛诺菲开发的SAR425899减弱了GCGR激动活性,从而规避了高血糖副作用,尽管早期数据表明它具有较好的血糖控制效果并有助于减轻体重,最终却因胃肠道副作用而止步于II期临床试验7;Cotadutide(MEDI0382)是阿斯利康开发的一款GLP-1R/GCGR双重激动剂,目前已经进入III期临床研究。然而IIb期临床结果显示,Cotadutide的降糖减重效果与GLP-1R单激动剂利拉鲁肽相近,加上一天一次的给药方式,令其在GLP-1R药物领域的竞争力难以凸显8,治疗2型糖尿病、肥胖症、糖尿病肾病和非酒精性脂肪性肝炎等适应症的开发被相继放弃。这些失败案例提示我们,GLP-1R/GCGR 双重受激动剂对两个受体的激动活性必须达到某种平衡,在增强疗效的同时规避相应的毒副作用。因此,通过三维结构解析阐明这些双重激动剂发挥药理作用的分子机制对进一步优化多肽序列具有重要意义。
王明伟/杨德华团队长期从事B1类GPCR的结构和功能研究,尤其关注基于GLP-1R的药物开发。继去年在Nature Communications上首次报道GLP-1R/GIPR双重激动剂替西帕肽和GLP-1R/GCGR/GIPR三重激动剂Peptide 20的配体识别和受体激活机制9后,他们又将视线转移至临床试验屡屡受挫的GLP-1R/GCGR双重激动剂。与内源性双重激动剂Oxyntomodulin相比,Peptide 15、SAR425899和MEDI0382对GLP-1R和GCGR具有不同的激动活性:Peptide 15对两者均有较强的激动作用,活性强于Oxyntomodulin并接近内源性配体;SAR425899对GLP-1R的激动活性比Oxyntomodulin强约10倍,而对GCGR的激动活性却减弱60倍;MEDI0382对GLP-1R的激动活性与Oxyntomodulin相近,但对GCGR的激动活性减弱了近5倍(图2A)。结构分析发现,这些配体的氨基端序列和识别模式基本相同,与受体产生较为保守的相互作用,而羧基端则决定了配体不同的药理活性和受体的特异性构象,尤其是引发GLP-1R的第一胞外环(Extracellular loop 1, ECL1)产生不同程度的偏移(图2B–C),进而改变了对GLP-1R的激动活性。比如MEDI0382的第18位氨基酸采用了精氨酸,而不是GLP-1的丙氨酸,导致ECL1的顶端发生轻微的向外偏转,从而改变了与周围氨基酸的相互作用模式,这可能是MEDI0382的GLP-1R激动活性弱于其他两者的原因。此外,结构信息还提示配体羧基末端的三个氨基酸对GCGR激动活性的调控至关重要:Peptide 15的羧基末端(M27-T29)与胰高血糖素相同,并与GCGR产生较强的相互作用,而SAR425899和MEDI0382的羧基末端更接近于GLP-1R激动剂的序列(图2B–D),可能是其GCGR激动活性较弱的原因。此前的研究结果同样显示,将Peptide 15的M27-T29替换成GLP-1的对应序列会大大减弱其GCGR激动活性并增强GLP-1R激动活性,进一步说明多肽羧基末端是调控双重激动活性的关键部位6。
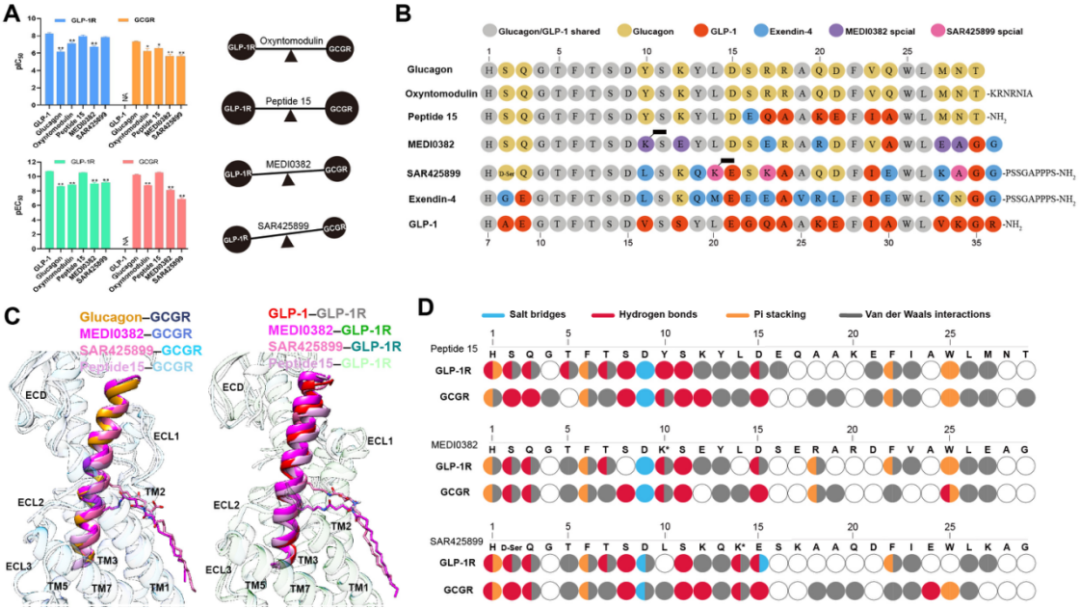
图2. GLP–1R/GCGR双重激动剂的药理活性和结构特征。A,GLP–1R/GCGR双重激动剂的受体结合和激动活性(左)以及与Oxyntomodulin受体激动活性的比较(右);B,GLP–1R/GCGR双重激动剂与内源性多肽的序列比对;C,GLP–1R/GCGR双重激动剂结合受体的结构特征。TM,跨膜螺旋(Transmembrane helix);ECL,胞外环(Extracellular loop);D,GLP–1R/GCGR双重激动剂与受体相互作用的指纹图谱。
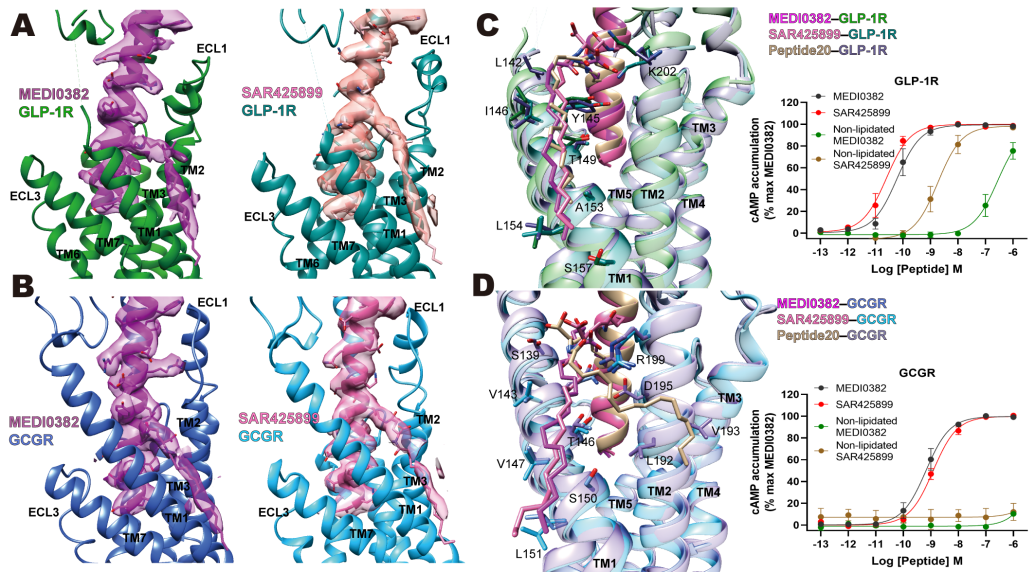
除了多肽序列上的差别外,脂链修饰也是调节双重药理活性的要素。MEDI0382和SAR425899的侧链修饰(即脂化的16碳棕榈酸)分别位于第10和14位的赖氨酸上,结合在两个受体的TM1和TM2之间,通过与受体的相互作用来稳定激活态构象(图3A-B)。采用化学方法去除脂链修饰后的MEDI0382和SAR425899诱导GLP-1R激活cAMP信号的能力分别下降了4,677倍和75倍,但都不再激活GCGR(图3C-D)。这不仅展现了侧链改造的药理学效应,而且验证了多肽与TM1-TM2的相互作用对GCGR的激活至关重要。
复旦大学基础医学院王明伟讲席教授、中国科学院上海药物研究所杨德华研究员和复旦大学基础医学院博士后丛朝彤研究助理为该论文的共同通讯作者;复旦大学基础医学院病原生物学系博士研究生李扬和丛朝彤为共同第一作者。合作者包括复旦大学基础医学院周庆同青年研究员、中国科学院上海药物研究所吴蓓丽研究员和复旦大学基础医学院应天雷研究员等。该项研究工作先后获得了国家自然科学基金委员会、国家卫生健康委员会、国家科学技术部、中国博士后科学基金和博新计划、海南省科技重大专项以及上海市科学技术委员会等机构的经费资助。
原文链接:
封面图来源:PNAS

本篇文章来源于微信公众号: 药时代
发布者:haitao.zhao,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 