


人类自然杀伤细胞 (NK) 占所有循环淋巴细胞的15%。NK细胞发现于20世纪70年代,主要与杀死感染的微生物和恶性转化的同种异体和自体细胞有关。NK细胞表现出抗肿瘤细胞毒性,无需事先致敏和产生细胞因子以及调节各种免疫反应的趋化因子。最近的研究表明,自然杀伤细胞的实际作用不仅限于杀死恶性转化和病毒感染的细胞,而且还直接或间接地影响适应性免疫系统的参与者,如树突状细胞和T细胞,扩展其功能域。此外,NK细胞亚群在其细胞毒性、细胞因子产生和归巢能力方面表现出主要的功能差异。根据细胞表面CD56的密度,将NK细胞分为CD56bright和CD56dim两者类型,它们具有不同的表型特征。CD56bright NK细胞具有产生丰富细胞因子的能力,而CD56dim NK细胞具有更强的细胞毒性,表达更多的免疫球蛋白样受体和FcγRIII(Fcγ受体III,又称CD16)。
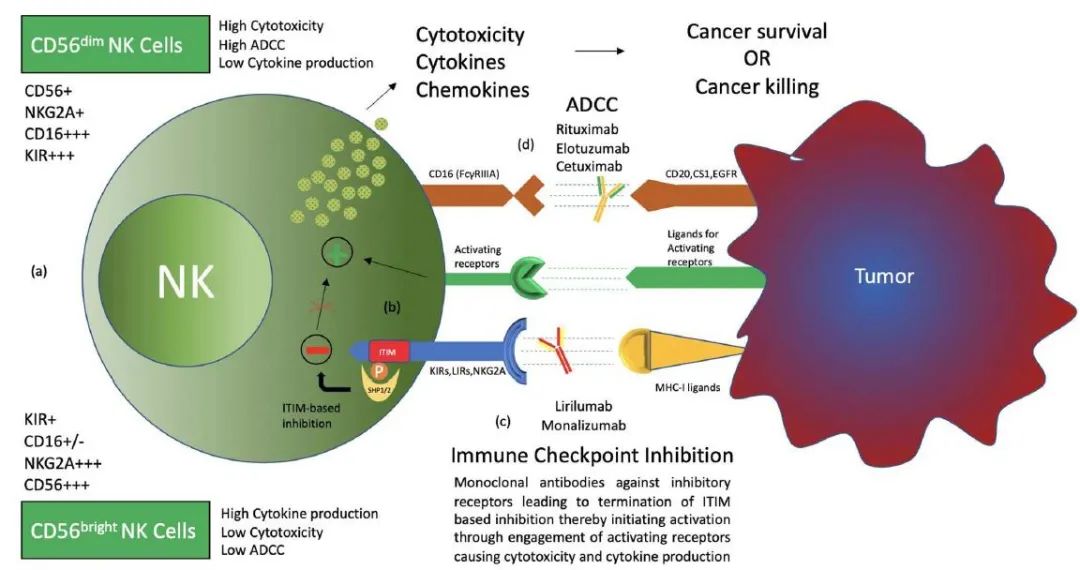
激活性受体和抑制性受体均在NK细胞表面表达,有助于NK细胞执行功能。MHC-I(主要组织相容性复合物Ⅰ类)抗原特异性的抑制性受体可密切调节NK细胞介导的细胞毒性和淋巴因子的产生。MHC-I特异性受体的抑制信号对于造血细胞避免NK细胞的破坏至关重要。这个概念被称为“丢失自我”,最初是由 Ljunggren和Karre 提出的。这种MHC-I识别抑制性受体形成了NK细胞表面受体的三个家族,即KIRs(杀伤细胞免疫球蛋白样受体)、LIRs(白细胞免疫球蛋白样受体)和NKG2A(自然杀伤细胞2族A)。KIRs是免疫球蛋白超家族的成员,是识别经典人类白细胞抗原A、B和C(HLA-Ia类)的I型跨膜分子。LIRs又称ILTs(免疫球蛋白样转录物),形成第二组受体,除了HLA Ia类外,主要识别非经典HLA-G(Ib类)分子。LIRs与KIRs属于同一个Ig超家族。NKG2A是NKG2族的一个成员,包括A、B、C、D、E、F和H,与CD94二聚形成NKG2A/CD94受体。它属于受体的C型凝集素家族,识别非经典HLA-EⅠ类分子作为其配体。
NK细胞的杀伤作用不仅需要通过抑制性受体检测转化细胞上的MHC-I分子,还需要通过激活性受体激活NK细胞。自然细胞毒性受体(NCR)是一组自然杀伤细胞表面激活性受体,包括NKp46、NKp30和NKp44。这些受体以及NKG2D和DNAM-1(DNAX辅助分子-1)识别病毒感染或恶性转化细胞表面表达的配体。一些共受体(2B4、NKp80、NTB-A和CD59)也被表达,它们只有与其他激活性受体结合才能发挥作用。CD16(或FcγRIII)也是一种激活性受体,主要由CD56dim NK细胞亚群表达,对IgG包被靶细胞的抗体依赖性细胞毒性(ADCC)至关重要。
肿瘤通过建立免疫抑制肿瘤微环境来逃避免疫系统。涉及NK细胞的免疫逃避包括几个机制。肿瘤细胞或肿瘤微环境的其他组分如转化生长因子-β(TGF-β)、IL-6、IL-10、色氨酸分解代谢产物、前列腺素E2(PGE2)、dickkopf相关蛋白2(DKK2)、吲哚胺2,3-双加氧酶(IDO)、可溶性HLA-G、可溶性NKG2D配体。据报道,半乳糖-3(NKp30可溶性抑制性受体)可降低NK细胞的活化、细胞毒性、IFNγ的产生及其激活性受体的表达和激活。此外,肿瘤细胞还发现激活性受体的配体脱落和抑制性受体配体的上调。因此,人们开发了多种策略来恢复NK细胞的功能,包括过继细胞转移、细胞因子治疗、靶向激活、抑制性受体和肿瘤微环境的单克隆抗体。肿瘤利用NK细胞抑制性受体进行免疫逃避就是这样一种机制,称为免疫检查点抑制,并已被证明是最有效和最受欢迎的治疗靶点。
KIR家族(也被称为CD158)是一类具多样性和多态性的NK细胞受体亚型,包含抑制性和激活性KIRs,每个受体识别特定的HLAⅠ类同系物(HLA-a、-B或-C)作为配体。抑制性KIR2DL1、KIR2DL2和KIR2DL3识别HLA-C作为它们的配体,而HLA-B和HLA-A作为其他KIRs的配体,包括抑制性KIR3DL1和KIR3DL2。除了NK细胞外,T细胞亚群和NKT细胞(不变自然杀伤性T细胞)也表达KIR。作为KIRs和KIRs配体的MHC-I分子(HLA-A、-B和-C)本身表现出广泛的自然多态性。KIR等位基因组合的多样性(染色体19q13,14上共有17个KIR基因),每个基因内的多态性,以及每个表达KIR的NK细胞,使得这个复杂的KIR序列能够识别MHC-I表达的微小变化。
IPH2101和lirilumab(IPH2102/BMS-986015)是针对KIR2DL1/2/3 NK细胞抑制受体的IgG4单克隆抗体,目前正在临床评估和开发中,作为单一疗法或与其他药物联合使用,包括分子靶向剂(来那度胺)、单克隆抗体(elotuzumab和rituximab)和免疫检查点阻断剂(ipilimumab和nivolumab)。IPH2101可用于各种血液学(AML、CLL、NHL)或实体恶性肿瘤(乳腺癌和卵巢癌)的临床评估。据报道,高达10mg/kg的IPH2101仍然具有良好的耐受性。到目前为止,IPH2101作为单一疗法在多发性骨髓瘤(MM)患者的临床应用中还没有取得令人满意的效果。
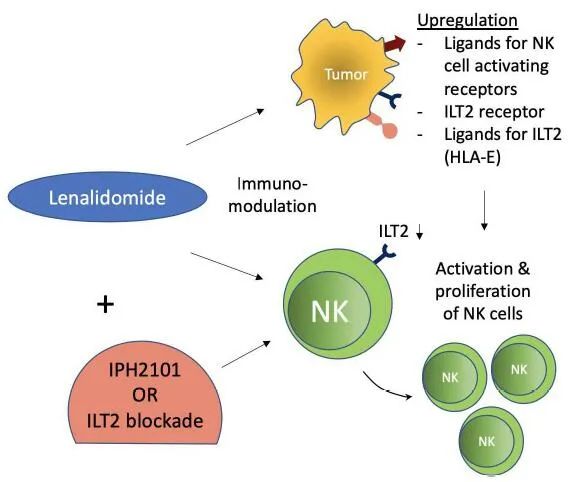
在RRMM中,IPH2101作为单药治疗的剂量递增I期试验,其主要目的是评估最大剂量耐受性和限制性毒性,报告了可接受的安全性和耐受性,但没有任何自身免疫的证据。然而,根据国际骨髓瘤工作组(IMWG)标准,只有11例(34%)的患者获得了疾病稳定的最佳反应。结果表明,在MM患者中输注IPH2101可降低NK细胞表面KIR2D受体的表达和NK细胞的反应性。临床前证据表明,来那度胺与IPH2101联用可增强NK细胞的功能,并上调NK细胞表面受体的配体,显示了对来那度胺耐药肿瘤的体内排斥作用。IPH2101和来那度胺作为MM患者的“双重免疫疗法”已经被报道达到了24个月的中位无进展生存率,5个具有可接受毒性的客观响应(5个严重不良事件),并且没有自身免疫。总的来说,尽管IPH2101不能作为单一的药物,但这种组合仍有希望对MM患者进行进一步的临床评估。
lirilumab的II期试验因未能达到为MM患者设定的客观疗效标准(M蛋白下降50%)而终止,总共9名入选患者中,仅有1名(11%)和6名(66%)达到最低反应和疾病稳定。然而,lirilumab增强了elotuzumab介导的细胞杀伤作用,并在KIR2DL3转基因和RAG缺陷小鼠中显示出增强抗肿瘤效果的协同作用。Sola等人也报道了lirilumab在体外增强了elotuzumab介导的ADCC作用以及lirilumab介导的有效的elotuzumab抗MM活性的体内协同作用,为MM患者的临床评估奠定了基础。目前正在进行一项I期(NCT2252263)研究,评估elotuzumab和lirilumab联合治疗多发性骨髓瘤患者的安全性。
体内IPH2101阻断KIR可提高存活率,临床前证据也表明对AML细胞(急性髓系白血病)有效。AML患者的临床疗效相对较好,平均PFS为7.7个月,RFS为10.8个月,OS为12.7个月。这些临床结果随着剂量的增加而改善,但程度不显著。与之前的0.3mg/kg剂量相比,只有在1-3mg/kg剂量下OS显著增加(27.9个月vs.11.8个月,p<0.034)。安全性和耐受性是可以接受的,不良事件均为轻微和短暂的。这项研究的延伸揭示了lirilumab作为老年患者维持治疗的作用,通过反复给药延长生存期和获得令人满意的安全性。然而,effiki试验的结果显示lirilumab与安慰剂组之间没有显著差异,这使得人们对lirilumab作为一种单一的药物产生了怀疑。lirilumab联合全剂量氮胞苷对重度预治疗/复发的AML患者具有良好的耐受性。最近的一项研究报告了lirilumab作为单药或联合阿扎胞苷治疗骨髓增生异常综合征(MDS)患者的有效性和耐受性。在人类模型中,与抗淋巴瘤治疗的细胞系模型相比,lirilumab和rituximab联合给药相对于rituximab单药显示出NK细胞毒性增加。在KIR转基因和同基因小鼠淋巴瘤模型中,这种组合在体外和体内都显示了NK细胞介导的细胞毒性增强,并依赖于rituximab。这些结果为临床评估抗KIR和抗CD20单抗的联合治疗提供了支持。
最近,这组抗KIR抗体的第三个成员IPH4102,一种人源化抗KIR3DL2单克隆抗体,已经进入临床评估阶段。IPH4102,也称为Lacutamab,在复发/难治性皮肤T细胞淋巴瘤的I期临床评估中耐受性良好,最常见的不良反应包括水肿、疲劳和淋巴细胞减少。临床活性也令人鼓舞,44例患者中有16例(36%)获得了总体疗效。复发/难治性皮肤T细胞淋巴瘤伴Sézary综合征的患者表现出更好的临床反应(43%)。一项II期临床试验(NCT03902184)正在研究IPH4102作为单一药物或联合化疗治疗T细胞淋巴瘤。
白细胞免疫球蛋白样受体(LIR)或免疫球蛋白样转录物(ILT)与KIR一样,属于Ig超家族,由激活性受体和抑制性受体组成。在总共11个LIR成员中鉴定出5个抑制受体(LIRB1-5)。许多免疫细胞(NK、T、B和包括巨噬细胞和树突状细胞在内的髓系细胞)不同程度地表达这些受体。其中,LIRB1(ILT2)和LIRB2(ILT4)除了其它配体外,还识别HAL-G作为它们的主要配体,从而导致免疫原性耐受。ILT2在自然杀伤细胞(正常NK细胞的36±18%)、T细胞、B细胞、单核细胞、树突状细胞亚群和髓源性抑制细胞(MDSCs)上表达,而ILT4主要在髓系细胞上表达。因此,ILT2和HLA-G相互作用可以抑制NK、T和B细胞的免疫功能,从而作为一个免疫治疗靶点。
各种原发性肿瘤和转移性恶性肿瘤都表达HLA-G,它也被认为是各种癌症进展和预后的指标。它的表达与各种癌症中NK功能的降低有关,如肝细胞癌(HCC)、卵巢癌、非小细胞肺癌(NSCLC)、胶质瘤和肾细胞癌(RCC)。此外,膜表面表达的HLA-G或可溶性HLA-G与ILT2的相互作用已证明抑制NK功能,包括细胞毒性、细胞因子产生和趋化因子分泌。蜕膜NK细胞的细胞溶解作用可以被HLA-G表达的靶细胞所抵抗。HLA-G的表达保护靶细胞免受外周血和NK细胞系中NK细胞毒性的影响。ILT2与HLA-G相互作用也抑制了靶细胞诱导的NK细胞产生极化IFN-γ。可溶性HLA-G(sHLA-G)对NK细胞(CD56bright和CD56dim)的趋化性、细胞因子和趋化因子的分泌有不同程度的调节作用。sHLA-G上调了CD56bright和CD56dimNK细胞CCL2的分泌,以及外周血CD56dim NK细胞CCL2、CCL8和CXCL2-CXCL3的分泌。HLA-G1/ILT2相互作用也通过抑制NK细胞毒性来减轻MICA/NKG2D的激活。
在胃癌中,HLA-G/ILT2相互作用可抑制浸润性NK细胞增殖和细胞毒性。然而,HLA-G在B细胞恶性肿瘤中有表达,由于B细胞恶性肿瘤也同时表达ILT2表面受体,因此反而抑制了恶性B细胞的增殖,而ILT2表面受体在实体瘤细胞中不表达。这可能是LIR-1阻断剂未能增强NK细胞对MM细胞的细胞毒性的原因。然而,LIR-1和NKG2A的双重阻断能够增加KIR阴性NK细胞的细胞毒性。这些发现提示HLA-G-ILT2相互作用起着双重作用:经典的抑制检查点在实体恶性肿瘤中的作用,以及由于ILT2在血液恶性肿瘤细胞上的表达而导致疾病进展的反作用。因此,用抗体阻断这个检查点可以被认为是实体癌的潜在靶点。需要进一步的研究来证实它在血液学癌症中的作用。来那度胺也显示了慢性淋巴细胞白血病(CLL)中NK细胞和白血病细胞的免疫调节作用。ILT2在CLL中的表达在NK细胞上增加,而在白血病细胞中表达降低。来那度胺的免疫调节作用增加白血病细胞ILT2的表达,并部分恢复其配体(HLA-E)的表达。此外,NK细胞活化和增殖也增加。ILT2阻断进一步增强NK细胞的活化和增殖。
NKG2A(也称为CD159)和CD94是C型凝集素家族的异二聚体抑制性受体,它识别非经典MHC-I分子HLA-E作为配体。CD94-NKG2A及其HLA-E配体呈非多态性。HLA-E*0101和HLA-E*0103是全球人群中仅有的两个HLA-E等位基因。外周血中近50%的NK细胞表达CD94/NKG2A,主要是那些不表达抑制性KIR的NK细胞。CD94/NKG2A与其他不同特异性的抑制性受体也存在共表达。此外,γδ和cd8+T细胞也表达CD94/NKG2A。NKG2A和CD94与正常细胞上表达的HLA-E反应可抑制信号激活,从而避免对正常旁观者细胞的破坏。
肿瘤细胞(血液学和实体瘤)为了避免NK细胞的杀伤,表现出HLA-E表达的上调。在各种癌症中,预后不良与HLA-E上调有关,包括结直肠癌、卵巢癌、妇科癌、肝癌、胶质母细胞瘤、霍奇金淋巴瘤、慢性淋巴细胞白血病、食管、胃、胰腺、结肠癌、肾脏、头颈部、肺癌和黑色素瘤。用抗体阻断CD94/NKG2A受体可作为一种治疗策略。因此,天然制药公司开发的抗CD94/NKG2A抗体(IPH2201 Monalizumab)已用于各种试验。体外和体内研究结果表明人源化抗NKG2A抗体对血液恶性肿瘤的应用是安全有效的。在体外实验中,monalizumab可改善慢性淋巴细胞白血病的NK细胞功能障碍。Monalizumab作为单药治疗妇科恶性肿瘤具有良好的耐受性(静脉给药或SC给药可达10mg/kg),未报告DTLs或SAEs。这项正在进行的大量预处理队列试验显示,41%的可评估患者(128人)病情稳定。
在免疫检查点抑制剂领域,从单一疗法向联合治疗方法的转变正在兴起,这主要是因为这些受体中的一些同时在一些先天性和适应性免疫细胞上大量表达,以及由于细胞间的相互作用和相互依赖性。Monalizumab正在与durvalumab、 cetuximab和ibrutinib联合评估。各种表达HLA-E的实体癌具有浸润性CD8+T、NK和NKG2A+免疫细胞。这些浸润的NKG2A+NK细胞和CD8+T细胞在受体阻断时表现出增强的NK和T细胞反应。已有报道PD-1与NKG2A在肿瘤浸润性NK细胞和CD8+T细胞中共表达。在体外和体内用抗体阻断NKG2A/HLA-E和PD-1/PD-L1通路均显示出完全缓解率。初步数据显示,monalizumab和durvalumab的联合应用已显示出临床疗效和可控制的毒性,且无DTLs,这是在大量预处理的转移性微卫星结直肠癌患者中发现的。
体外研究发现,抗NKG2A抗体与其他肿瘤免疫疗法具有协同效应。monalizumab联合cetuximab对先前治疗过、复发和/或转移的头颈部鳞状细胞癌(SCC)的安全性和有效性进行了初步评估,结果显示联合用药的ORR(客观缓解率)为27.5%,5个月的中位PFS(无进展生存率)和10个月的中位总生存率(OS)。如果与以往研究中单用cetuximab疗效的历史记录相比,这是一个令人鼓舞的结果(ORR12.6%,PFS 2.3 m,OS 5.6 m)。联合治疗的不良反应与单用cetuximab相似。最近的体内分析表明,NKG2A对CD8+T细胞的诱导会阻碍疫苗治疗的效果,阻断NKG2A受体可提高疫苗治疗的疗效。总的来说,阻断NKG2A代表了一种令人兴奋的治疗方法,特别是,它与其他免疫肿瘤治疗药物的结合是前进的方向,并值得进一步的探索。
TIGIT(具有免疫球蛋白和ITIM域的T细胞免疫受体)是一种在NK和T细胞上表达的免疫抑制性受体,如活化的NK、T、mT(记忆T细胞)、fTh(滤泡辅助性T细胞)和调节性T细胞(Tregs)。与TIGIT相比,CD96是同一免疫球蛋白超家族的成员,具有类似的抑制作用,但与配体CD155的结合亲和力较低。CD226是一种激活性受体,与TIGIT和CD96竞争结合CD155。CD155(主要)和CD112作为TIGIT和CD96结合的配体,以抑制T细胞和NK细胞介导的免疫。CD155是一种跨膜糖蛋白,又称脊髓灰质炎病毒受体(PVR),最初被鉴定为脊髓灰质炎病毒进入受体。CD155是免疫球蛋白超家族的一员,也是连接蛋白样分子家族的第五个成员,因此也被称为necl-5。它在正常人体组织中几乎不表达,但许多肿瘤细胞系和原发性恶性肿瘤高表达CD155。在CD155的功能中,通过其与抑制性受体TIGIT和CD96以及激活受体CD226的相互作用来进行免疫调节是特别令人感兴趣的。各种癌症都表现出CD155的上调,相应的TIGIT和CD96的NK和T细胞表达上调,以通过诱导T细胞或NK细胞抑制来逃避抗肿瘤免疫。临床前证据支持阻断NK细胞介导的抗肿瘤免疫激活检查点的想法,临床转化正处于初级阶段。
NK、效应和记忆T细胞和调节性T细胞表达TIGIT。到目前为止,TIGIT阻断主要是在血液肿瘤中得到评估,TIGIT阻断多发性骨髓瘤主要与CD8+T细胞有关。多发性骨髓瘤细胞CD155配体上调,其免疫监视和治疗依赖于NK和T细胞分泌的CD226。阻断TIGIT阻止了干细胞移植后骨髓瘤逃逸的T细胞衰竭机制。CD8+T细胞表面表达高水平TIGIT与多发性骨髓瘤进展相关,通过在小鼠和人类的多发性骨髓瘤中阻断TIGHT,增强了对多发性骨髓瘤的免疫反应。在PD-1靶向药物治疗无效的情况下,抗TIGIT治疗可作为单一治疗或与其他治疗药物联合治疗多发性骨髓瘤患者,其临床疗效已在进行测试。
急性髓系白血病(AML)患者不良的临床预后和CD8+T细胞衰竭与TIGIT相关。通过体外单独阻断TIGIT和PVR或PVRL2相互作用或结合BiTE®双抗AMG 330,T细胞显著增强了AML细胞的溶解。同种异体移植后骨髓中TIGIT表达较高的患者,其II-IV级急性移植物抗宿主病(aGVHD)的发生率显著降低(p=0.048),PFS缩短(p=0.024),OS缩短(p=0.046)。TIGIT的高表达也降低了同种异体移植后BM中NK细胞的数量,提示TIGIT可能在GVL效应和GVHD中起重要作用,从而控制同种异体移植后NK细胞的活性和增殖。基于这些观察结果,提示TIGIT可能是异基因造血干细胞移植后的预后预测因子,阻断TIGIT可能是一种有效的免疫治疗策略,可以增强AML患者异基因造血干细胞移植后移植物对白血病的影响。Hodgkin和Reed-Sternberg(HRS)细胞或Tregs通过PD-1参与Th1、CD8+T细胞和NK细胞活性的抑制。在霍奇金淋巴瘤患者CD3+T细胞上观察到PD-1和TIGIT的可变表达,提示TIGIT阻断剂单独或联合其他药物可能是一个潜在的治疗靶点。然而,还需要进一步评估。由于PD-1或TIM-3由TIGIT阳性T细胞共同表达,靶向TIGIT可能是避免B细胞非霍奇金淋巴瘤(B-NHL)中T细胞耗竭的另一种机制。虽然HL和NHL在T细胞上表达TIGIT,但其在NK细胞上的表达及在这类患者中的相关治疗应用至今尚未得到评价。
TIGIT固有表达抑制NK和CD8+T细胞功能,从而帮助肿瘤(结肠直肠癌)在体内生长。TIGIT与荷瘤小鼠和结肠癌患者的NK细胞耗竭有关,而这种耗竭通过其阻断而恢复,从而激发了强大的抗肿瘤免疫。NK细胞的存在对于TIGIT和/或PD-L1阻断或双重阻断这两个检查点的治疗效果非常重要,因为NK细胞缺失与IFNy-或TNF-分泌TIL(CD8+)频率较低和PD-1表达TIL(CD8+)频率较高相关。NK细胞占肝脏淋巴细胞的25-50%,这表明它们对肝脏免疫的重要性。此外,HCC患者的生存和预后与血液和肿瘤组织中的NK细胞数呈正相关。HCC患者的肿瘤进展与肿瘤浸润性NK细胞功能紊乱有关,主要是CD11b-CD27-NK亚群。Sun等人发现衰竭的肿瘤浸润性CD96+NK细胞,发现其表达与HCC患者不良临床结局相关。当CD96-CD155相互作用或TGF-β1被阻断时,NK细胞衰竭被逆转。
近年来,检查点抑制剂的联合应用越来越受到重视,以达到协同效应。据报道,通过增强CD8+T细胞活化,PD-1和TIGIT双重靶向的荷瘤小鼠的生存率得到了提高。Dixon等人报道,在MC38结肠癌模型中,TIGIT和PD-1双重阻断可产生协同抗肿瘤作用,导致肿瘤完全消退。在黑色素瘤患者中,TIGIT和PD-1双重阻断可协同增加肿瘤浸润和肿瘤抗原特异性CD8+T细胞的增殖、脱颗粒和细胞因子分泌,显示双重阻断的可能性。Hong等人提示PD-1和TIGIT也可以作为治疗RCC的潜在靶点。在GBM患者中,这种双重阻滞剂还可以提高抗肿瘤免疫力和生存率。虽然这些研究通过探索T细胞的作用来反映双检查点阻断在各种癌症中的疗效,但也有一些研究表明双检查点的疗效也依赖于NK细胞。抗TIGIT加抗PD-L1阻断剂可防止荷瘤小鼠和结肠癌患者的NK细胞衰竭。另一方面,抗CD96联合阿霉素化疗、抗CTLA-4或抗PD-1在三种不同的肿瘤模型中显示出更有效的抑制肿瘤转移。膀胱癌(BC)患者的衰竭NK细胞在外周和肿瘤中均显示TIM-3和TIGIT的上调。事实上,TIGIT和CD96在各种癌症中NK细胞衰竭中的作用仍在研究中,还需要进一步的揭示,以确定其作为单药治疗或与其他检查点联合使用的潜力。
唾液酸结合免疫球蛋白样凝集素(Siglecs)是一种免疫调节性唾液酸结合受体,属于I型凝集素家族。Siglecs在各种免疫细胞上表达,这些免疫细胞包括淋巴和髓系来源的免疫细胞,即中性粒细胞、嗜酸性粒细胞、单核细胞、巨噬细胞、NK细胞、树突状细胞、肥大细胞以及B和T细胞。Siglecs在两个性质上表现出多样性:表达和对含唾液酸配体的特异性。这些Siglec大多是抑制性受体,如Siglec-2、Siglec-3、Siglec-5、Siglec-6、Siglec-7、Siglec-8、Siglec-9、Siglec-10和Siglec-11。在抑制性Siglec中,Siglec-7和Siglec-9被报道在人类NK细胞上表达。与经典NK细胞抑制受体NKG2A/CD94和KIRs类似,抑制性siglec在其胞内段C端也含有一个或多个ITIM和ITIM样基序。连接后,ITIM被Src家族激酶磷酸化,招募并激活Src同源2(SH2)结构域的蛋白质,主要是酪氨酸磷酸酶SHP1和SHP2或细胞因子信号转导3蛋白的抑制因子(SOCS3)。
唾液酸(一种九碳糖)的变化与癌症有关。除了异常的肿瘤细胞表面表达外,唾液酸的改变和唾液酸的含量或密度与肿瘤的发生和发展有关。这些包括高唾液酸化、异种唾液酸(摄取Neu5Gc)和唾液酸改变,包括唾液酸的C5羟基修饰(产生KDN)和唾液酸的O-乙酰化(特别是9-O-乙酰化)。高唾液酸化与一些癌症有关,如口腔癌、肾细胞癌、头颈部鳞状细胞癌、乳腺癌、前列腺癌和结肠癌。KDN的变化与卵巢癌和头颈部癌有关,而唾液酸的O-乙酰化在结直肠癌中有报道。
Siglec-唾液酸相互作用参与免疫耐受的调节,并可作为诱导抗肿瘤免疫的靶点。针对这些抑制检查点抗体(抗Siglec-2;Inotuzumab ozogamicin和抗Siglec-3;Gemtuzumab ozogamicin)与细胞毒性剂结合的抗体偶联药物,已经在临床测试了疗效。人类NK细胞主要上调Siglec-7和Siglec-9。此外,在癌症中,外周NK细胞也上调Siglec-9,主要在cd56dimcd16+NK细胞上。Fab片段阻断Siglec-7和Siglec-9可提高NK细胞体外抗肿瘤细胞(K562)的细胞毒性。在移植人NK细胞和人肿瘤细胞的免疫缺陷小鼠体内模型中,肿瘤细胞的杀伤是通过抑制NK细胞的唾液聚糖依赖性介导的。已开发的NK-92MI细胞系的Siglec-7阴性表型表明对白血病细胞具有高度和持续的细胞毒性。唾液酸酶治疗后,乳腺、脑、结肠、肝脏或淋巴组织的各种重塑肿瘤株(Siglec-7丰富型肿瘤细胞系)对NK细胞杀伤的敏感性增加。唾液酸酶与靶向HER2抗体的体外融合增强了NK细胞对HER2+肿瘤细胞的杀伤作用。通过唾液酸酶切断唾液酸配体,特别是Siglec-7和Siglec-9结合的配体,可以增强NK细胞介导的杀伤作用。这表明,这种抗体唾液酸酶结合物使肿瘤细胞的表面糖蛋白选择性去唾液酸化可以使肿瘤更容易受到的ADCC的影响。
高亲和力Siglec-9抗体通过阻断唾液酸在肿瘤靶细胞上的表达而增强NK细胞的细胞毒性。这些针对Siglec-9的抗体也提高了NKG2A阻断剂诱导的抗肿瘤反应。Siglec-9在非小细胞肺癌、卵巢癌和结直肠癌中对肿瘤浸润的CD8+T细胞上调。黑色素瘤中的肿瘤内效应记忆CD8+T细胞亚群也显示Siglec-9通过磷酸化SHP1参与上调和抑制。体外和体内靶向唾液酸聚糖-SAMP/Siglec通路可增强抗肿瘤免疫。其他抑制性受体如PD-1也由表达Siglec-9的T细胞共同表达,这提示了共同抑制的可能性。Siglec-9在不同类型的免疫细胞上表达,提示Siglec-9的多模式作用。这些数据支持了这样一种观点,即抗Siglec-7和抗Siglec-9阻断抗体可以开发用于癌症免疫治疗,并可与其他免疫检查点抑制剂结合使用。
LAG-3(淋巴细胞活化基因-3)也是免疫球蛋白超家族受体的一员,具有抑制性。LAG-3被发现在活化的CD4+T细胞、CD8+T细胞和NK细胞表面上调。除了这些细胞外,Lag-3还表达在其他几种免疫细胞上,包括TIL、调节性T细胞、iNKT细胞、B细胞和DC细胞。它识别MHCII类分子,与CD4分子结构相似,但与MHC-II分子结合的亲和力大于CD4。LSECtin在肝脏和其他几种肿瘤中表达,是DC-SIGN家族的一员,也被描述为表达LAG-3免疫细胞的潜在配体。LAG-3参与抑制T细胞效应器功能,并参与T细胞衰竭。它还促进调节性T细胞的抑制活性。阻断LAG-3已被证明可以诱导T细胞功能的改善。Relatlimab是一种抗LAG-3单克隆抗体,目前正在进行的几项临床试验中进行研究,无论是单独使用还是与PD-1阻断剂联合应用,都可以用于各种癌症。LAG-3和PD-1在T细胞功能调节中显示出协同作用,以促进肿瘤免疫逃逸。
尽管LAG-3在NK细胞上有表达,但其在NK细胞调控中的作用尚未得到充分证实。在小鼠模型中敲除LAG-3基因导致NK细胞不能杀死某些肿瘤靶点。然而,这种缺失对MHC I类错配的细胞溶解活性没有影响。另一方面,人类NK细胞却表现出相反的结果。阻断LAG-3通路的抗体不能诱导人NK细胞产生细胞毒性。可溶性LAG-3能与MHC-II分子结合,对人类NK细胞的杀伤能力也没有影响。在HIV患者中,病毒控制与NK细胞上LAG-3的低表达以及其他抑制分子有关。Wiskott-Aldrich综合征蛋白(WASp)缺乏与癌症的高易感性相关,很可能是由于NK细胞和DC的抗癌能力受损所致。WASp敲除的NK细胞显示出细胞衰竭和NK细胞记忆LAG-3表达增强相关。这似乎有一个明显的关联,然而,LAG-3对NK细胞功能的直接影响和潜在机制还需要进一步研究。与NK细胞相比,其对NKT(自然杀伤性T细胞)功能的调节已被广泛报道。在慢性HIV患者中,iNKT细胞耗竭和IFN-γ产生减少与LAG-3表达升高有关。LAG-3信号通路通过阻断S期细胞周期,下调活化的CD1d限制NKT细胞的增殖。
可溶性重组LAG-3-Ig融合蛋白Eftilagimod alpha(IMP321)已被用作免疫佐剂,用于预防各种感染和癌症。它也被应用于癌症的单一治疗或联合化疗。在体外的短期试验中,IMP321能够在健康个体(60个捐赠者中的52个)诱导NK细胞产生细胞因子(IFN-γ和/或TNF-α),以及在较低程度上在21个未经治疗的转移性癌症患者体内诱导产生细胞因子。在转移性肾癌患者中,IMP321在剂量递增研究(P003)中,作为单药治疗诱导NK细胞活化。标准化疗的IMP321与乳腺癌患者数月内NK细胞活化增强相关。因此,LAG-3具有激活T细胞和NK细胞的潜力。因此,它可以作为检查点抑制的潜在靶点进行进一步的研究。此外,在最近对CIML NK细胞的研究中,CD56bright、CD16-和CD62L+NK细胞被鉴定为细胞因子诱导的记忆样(CIML)NK细胞的优势亚群,NKG2A的持续表达参与抑制HLA-E阳性靶细胞的杀伤。CIML-NK细胞亚群KIR+和NKG2C+表达LAG-3,表明CIML-NK细胞是双重检查点抑制的潜在靶点。
共抑制受体TIM-3(T细胞免疫球蛋白和粘蛋白结构域3)识别galectin-9作为配体,在各种癌症和慢性感染中上调。此外,TIM-3可变IgV结构域也被报道与其他配体结合,如HMGB1(高迁移率组蛋白B1蛋白)、Ceacam-1(癌胚抗原细胞粘附分子1)和PtdSer(磷脂酰丝氨酸)。TIM-3的表达是多种多样的,包括几种类型的免疫细胞,包括CD4+T细胞、CD8+T细胞、调节性T细胞、B细胞、NK细胞、NKT细胞和髓样细胞。TIM-3与其配体的结合通过耗尽T细胞和NK细胞来诱导免疫耐受。这种途径的上调与各种慢性感染和癌症中T和NK细胞的耗竭有关,使TIM-3成为T和NK细胞免疫的负调节因子。相应地,它的阻断作用逆转了T细胞或NK细胞功能障碍。TIM-3和PD-1的共表达参与介导了多种癌症和慢性病毒感染中CD8+T细胞衰竭。研究表明,TIM-3和/或PD-1阻断可逆转T细胞耗竭,并减缓肿瘤生长。针对TIM-3的抗体,如Sym023、Cobolimab、LY3321367、BGB-A425和MBG453,以及几种抗PD-1/PD-L1抗体,正在进行临床研究,以确定其对各种癌症的疗效。
TIM-3在NK细胞上的表达有几个方面的原因。它被认为是成熟、活化和预后的标志物。与CD56+/CD3+NKT和CD56-/CD3+T细胞群相比,TIM-3在静止CD56+/CD3+NK细胞群中高表达。健康成人血液中部分成熟CD56dimCD16+NK细胞亚群显示TIM-3表达,而其在未成熟CD56brightCD16-NK细胞亚群中的表达不均一。此外,几种细胞因子(IL-12、IL-15和IL-18)强烈诱导TIM-3的表达,主要是在未成熟的CD56bright NK细胞中。IL-12和IL-18诱导的NK细胞活化和IL-15诱导的成熟是TIM-3在这些细胞中表达的主要原因,识别出TIM-3在NK细胞中的表达作为活化和分化或两者兼而有之的标记。TIM-3在外周NK细胞中的上调在一些癌症中被观察到,即胃癌、肺腺癌、晚期黑色素瘤和膀胱癌,导致NK细胞衰竭。肿瘤生长的NK细胞中TIM-3水平的增加表明TIM-3的表达是一个预后的生物标志物。GIST(胃肠道间质瘤)和膀胱癌患者的肿瘤浸润性NK细胞也表达TIM-3。与TIM-3和PD-1在T细胞中的共表达相似,衰竭的肿瘤浸润NK细胞在MHC-I缺陷肿瘤中也显示出可检测的共表达。然而,GIST患者在TIM-3+肿瘤浸润性NK细胞中缺乏PD-1共表达。
CD200R是另一种在T、B、NK和髓系细胞上表达的抑制性受体。它识别CD200作为其配体,除了在各种肿瘤上表达外,CD200还表达于各种正常组织,如中枢神经系统、视网膜、毛囊细胞、血管内皮细胞和胸腺细胞,以及活化的T、B和DC细胞。CD200被认为是肿瘤进展的标志物,因为它在造血和非造血源的各种癌症上过度表达,如急性髓系白血病、多发性骨髓瘤、毛细胞白血病、B细胞慢性白血病、黑色素瘤、卵巢癌、直肠癌和膀胱癌,其表达与最差预后相关。此外,CD200的表达也可以诱导癌细胞。事实上,癌细胞表达CD200对CD200–CD200R信号抑制抗癌反应没有影响。因此,CD200阻断剂是一种潜在的治疗选择,不局限于治疗表达CD200的肿瘤。抑制性CD200-CD200R通路通过调节巨噬细胞和DC细胞间接抑制T细胞的效应器功能。因此,阻断CD200–CD200R的相互作用可以抑制肿瘤生长,这支持了拮抗性CD200或CD200R抗体作为癌症治疗的一种选择。Samalizumab(人源化抗CD200抗体)耐受性良好,CD4阳性T细胞和CD200阳性B-CLL呈剂量依赖性变化,Th1细胞因子反应中等。
对于NK细胞,有证据表明CD200–CD200R抑制途径直接参与了NK细胞的抑制。在AML患者中,CD200的过度表达抑制了NK细胞的抗肿瘤反应,从而增加了这些患者复发的风险。AML患者的NK细胞亚群表达CD200R,提示CD200-CD200R相互作用抑制了NK细胞。此外,在CD200hi患者中,阻断CD200的抗体可恢复NK细胞的活性。这些数据表明CD200-CD200R相互作用直接导致AML患者NK细胞的抑制。这是唯一直接表明表达CD200的靶细胞能够抑制NK细胞的细胞毒性和IFNγ产生活性的研究。Liu等人在研究CD200+黑色素瘤生长和转移限制中的CD200信号部分时发现,在CD200缺乏的转移性肿瘤生长的肝脏中,NK细胞的数量显著减少。然而,CD200缺乏如何影响肝脏局部NK反应仍有待解释。这提示CD200-CD200R检查点是血液学和实体瘤检查点阻断的一个很好的靶点。由于NK细胞与AML和多发性骨髓瘤中的其他检查点受体(如KIR和NKG2A)相关,因此在这类患者中可以验证联合检查点靶点。
CD47,也被称为整合素相关蛋白(IAP),是免疫球蛋白超家族中广泛表达的糖蛋白。其作为参与β3整合素介导的信号转导的白细胞膜蛋白被首次发现。它是一种跨膜蛋白,除了整合素外,还与血小板反应蛋白-1(TSP-1)和信号调节蛋白α(SIRPα)相互作用。在CD47所执行的功能中,它与SIRPα和TSP-1的结合,确立了其作为一种抑制受体的作用,通过抑制吞噬、抗原呈递和T/NK细胞抑制参与癌症的免疫逃避。因此,用抗体靶向这个信号通路显示出潜在的治疗肿瘤的能力。
CD-47在NK细胞介导的抗病毒或抗肿瘤细胞毒性中起抑制作用。NK细胞毒性与HNSCC细胞CD47表达有关。高表达CD47的HNSCC细胞株表现出较低水平的NK细胞毒性。用中和MHC-1或抗CD47抗体预处理细胞可增加NK细胞对HNSCC细胞系的细胞毒性。SIRPα和TSP-1均与NK细胞介导的细胞毒性有关。在免疫功能正常的同基因小鼠中,抗SIRPα抗体通过阻断CD47的相互作用而显著抑制RCC或黑色素瘤细胞的肿瘤形成。除了巨噬细胞和CD8+T细胞外,NK细胞的选择性耗竭大大削弱了抗SIRPαAb的抗肿瘤作用。然而,在体外,NK细胞对肿瘤细胞的杀伤作用不受同一抗体的抑制,提示NK细胞的CD47功能与SIRPα无关。与此类似,TSP-1也被证明在抑制NK细胞早期增殖和促进晚期扩张中发挥作用,但CD47的作用尚未被证实。因此,在这方面仍有许多问题有待解决。然而,用抗体攻击CD-47是值得探索的,不仅在巨噬细胞、树突状细胞和T细胞,而且在NK细胞。CD47与PD-L1的双重阻断作用也已被研究,并被证明能增强对循环肿瘤细胞的免疫治疗作用。抗CD47抗体Magrolimab(Hu5F9-G4)正在多个I期和II期临床试验中与其他药物(如利妥昔单抗、西妥昔单抗、阿扎胞苷、acalabrutinib和atezolizumab等)进行不同组合的研究。
CTLA-4(细胞毒性T淋巴细胞相关抗原4)是一种共抑制受体,可在多种免疫细胞上表达,如荷瘤小鼠的活化T淋巴细胞(CD4+T细胞和CD8+T细胞)、调节性T细胞、肿瘤浸润性NK细胞和脾脏Kit+CD11b−NK细胞,并在IL-2刺激下诱导小鼠NK细胞。CTLA-4与共刺激受体CD28竞争癌细胞或抗原呈递细胞上的配体B7-1(CD80)和B7-2(CD86)。CTLA-4被公认为T细胞活化的负调节因子和外周T细胞耐受性和自身反应性的调控者。用抗体阻断CTLA-4(ipilimumab)可以改善各种癌症的T细胞功能。
已有研究表明CTLA-4/CD28/CD80/CD86通路参与NK细胞介导的细胞毒性。在体内,CD28触发NK细胞的增殖,它们的细胞毒性和细胞因子的分泌已经在许多研究中被描述。癌细胞上的配体B7-1和B7-2似乎也能提高人类NK细胞的细胞毒性。同样,NK细胞表达的CTLA-4作用树突状细胞的B7-1可以抑制IFN-γ的产生。在小鼠中,肿瘤细胞产生的IL-18可诱导具有B7-H1依赖性免疫清除功能的Kit+CD11b−NK细胞。Kit+CD11b−NK细胞也报告CTLA-4上调;然而,其在NK细胞控制的癌症中参与肿瘤进展的情况尚未被研究。这些研究揭示了这一途径在NK细胞介导的细胞毒性中的不可否认的作用。然而,人类NK细胞中不存在CD80-CD28/CTLA-4介导的共刺激。共刺激CD28/B7也被否认在小鼠CMV感染的外周NK细胞中起任何重要作用。
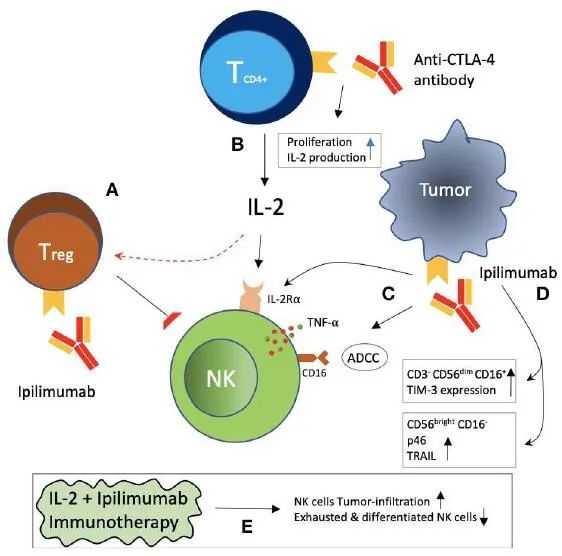
CTLA-4+肿瘤浸润性NK细胞是一个基于抗CTLA-4单克隆抗体的前瞻性免疫治疗靶点。如上图所示,阻断CTLA-4可能间接地减轻被抑制的NK细胞。CTLA-4在Tregs上的表达被认为是其抑制功能所必需的。在西妥昔单抗治疗的头颈部癌患者中,NK细胞毒性抑制和CTLA-4阳性Tregs增加与预后不良相关。Ipilimumab是一种抗CTLA-4单克隆抗体,导致Tregs的耗竭,以Fc介导的方式对黑色素瘤患者产生临床疗效,部分原因可能是由于减轻了Tregs对NK细胞毒性的抑制。Ipilimumab还可以通过原代NK细胞以及IL-2激活的NK细胞和γδT细胞上的FcyRIIIA与黑色素瘤细胞系和组织上的CTLA-4反应来触发ADCC作用。此外,Ipilimumab和CTLA-4阳性黑色素瘤细胞的相互作用也导致NK细胞释放TNF-α。CTLA-4阻断和IL-2免疫治疗联合应用可延缓黑色素瘤的生长和延长生存期,显示协同作用。CTLA-4阻断可增加免疫细胞(包括CD8阳性T细胞和NK细胞)的肿瘤浸润,而IL-2可降低肿瘤浸润性NK细胞耗尽和分化的比例。Ipilimumab在NK细胞表型上诱导IL-2Rα链表达,随后增强对IL-2刺激和细胞毒性的反应,这与晚期黑色素瘤患者更好的临床反应相关。
程序性死亡受体-1(PD-1)在各种免疫细胞上表达,包括T细胞(CD4+&CD8+)、B细胞和髓细胞、NK细胞、NKT细胞和其他固有淋巴细胞。PD-1、PD-L1和PD-L2配体的上调在各种癌症中已有报道,它们的相互作用导致T细胞抑制,导致免疫逃逸。大约四分之一健康人的外周血中可检测到NK细胞上PD-1的高表达。然而,在癌症患者,如卵巢癌患者的腹水、卡波西肉瘤患者的外周血、肾细胞癌和多发性骨髓瘤患者中,NK细胞上PD-1的表达上调。在消化道癌,如食道癌、胃癌、胆管癌、肝癌和结直肠癌中,外周血NK细胞和肿瘤浸润性NK细胞也同样上调。慢性感染如HIV(人类免疫缺陷病毒)、HCV(丙型肝炎病毒)、HCMV(人类巨细胞病毒)和结核分枝杆菌也显示出NK细胞PD-1表达增强。
PD-1在NK细胞上的表达是多种多样的,因癌症而异。PD-1在CD56bright NK细胞中普遍缺乏表达。然而,CD56dimNK细胞已证明PD-1表达受限于NKG2A−KIR+CD57+表型,一种完全成熟的NK细胞。NKG2A−KIR+CD57+表型NK细胞被认为具有显著下调的激活受体,如NKp30和NKp46。此外,PD-1表达与NK细胞抗肿瘤活性受损之间存在相关性,而抗体干扰PD-1和PD-L1的相互作用导致部分修复。PTLD儿童移植患者也显示出NK细胞功能改变,PD-1增加,NKp46和NKG2D表达降低。另一方面,CD56bright NK细胞在慢性HCV患者中表达PD-1。同时,在消化道癌症患者中,两种类型的NK细胞(CD56bright 和CD56dimNK细胞)都显示PD-1表达增加。此外,新发现的肝癌组织中浸润的CD3−CD49a+CD56+NK细胞也显示其表面有大量PD-1表达,这与肝细胞癌患者的生存率降低有关。
在一些癌症中,NK细胞PD-1表达上调表明NK细胞处于功能失调状态,可能是由于缺乏MHC-I的肿瘤细胞过度刺激所致。比较PD-1+NK细胞和PD-1-NK细胞发现PD-1+NK细胞功能衰竭,细胞毒性和细胞因子生成受损,增殖能力降低。抗PD-1单抗的阻断已被证明能恢复NK细胞的功能。小鼠肿瘤细胞表达PD-1,抗PD-1阻断剂诱导NK细胞产生抗肿瘤免疫应答。在体外,抗PD-1抗体增强了NK细胞介导的对自体MM细胞的杀伤作用。PD-1阻断剂也促进了小鼠NK细胞对小鼠胶质瘤干细胞的杀伤作用。
B7同源物3蛋白(B7-H3)是B7-CD28家族的配体分子,其受体可能存在于T细胞和NK细胞上,它似乎可以同时抑制T细胞和NK细胞的功能,但尚未被证实。B7-H3被认为可以同时共刺激和共抑制来调节T细胞功能。B7-H3通过与TLT-2结合来刺激T细胞活化,而与未知受体的结合导致T细胞的共同抑制。同时,通过激活未知受体,它可以抑制NK细胞和成骨细胞。
B7-H3在胰腺、肝脏、小肠、结肠、心脏、胸腺、脾脏、胎盘和睾丸等各种正常组织上的表达有限。然而,B7-H3的异常表达可见于各种与预后不良相关的恶性肿瘤,包括肾细胞癌、乳腺癌、肺癌、食管鳞癌、胃癌、胰腺癌、胆囊癌、结直肠癌、前列腺癌、卵巢癌、宫颈癌、子宫内膜癌、骨肉瘤,神经母细胞瘤。肺癌、肾细胞癌、肝细胞癌、结直肠癌和胶质瘤患者的循环血清B7-H3水平显著高于健康志愿者。抑制NK细胞介导的细胞毒性是B7-H3表达细胞逃避肿瘤的多种机制之一。胶质瘤的恶性程度和生存率降低与肿瘤和内皮细胞中B7-H3的表达有关。胶质瘤细胞上清液中可溶性B7-H3和细胞结合B7-H3均能抑制自然杀伤细胞介导的肿瘤细胞溶解。在B7-H3沉默的胶质瘤细胞系的体内模型中证实了对杀伤的敏感性。单克隆抗体介导的4Ig-B7-H3分子被鉴定为神经母细胞瘤相关分子,对细胞转染物或新分离的神经母细胞瘤细胞进行掩蔽,保护其不被NK细胞杀死。类似地,在卵巢囊性畸胎瘤中的神经母细胞瘤中,除了大量的HLA-I类分子外还表达B7-H3,这表明神经母细胞瘤细胞对NK细胞介导的溶解具有保护性的免疫逃避机制。此外,与来自外周血的NK细胞相比,这些患者腹腔液分离的NK细胞上DNAM-1(CD226)和CD16等受体的表达强度较低。以B7-H3作为靶点的BiKE双特异性抗体在治疗NSCLC时,可以通过诱导自然杀伤细胞显著抑制肿瘤细胞生长。
B7-H3结合Fc优化的人源化IgG1单克隆抗体,Enoblituzumab,目前正在探索中。它已经被证明可以抑制B7-H3阳性的肾和膀胱癌移植物的肿瘤生长。MGA271是一种Fc优化的靶向B7-H3的人源化单克隆抗体,已在几种肿瘤类型中显示出安全性和抗肿瘤疗效。这种抗肿瘤活性归因于患者T细胞克隆性的增加。Enoblituzumab的进一步鉴定,包括其药理学动力学和动力学,以及其安全性、剂量耐受性和抗肿瘤活性,以对抗年轻患者中B7-H3受体阳性表达的复发或难治性实体恶性肿瘤,正在一项开放标签I期研究(NCT02982941)中进行评估。Orlotamab(MGD009,一种人源化B7-H3 x CD3 DART®蛋白),是一种除B7-H3外靶向CD3的双特异性抗体,由Macrgenenics开发,目前正在进行临床研究(NCT03406949),评估该抗体与抗PD-1抗体(MGA012)联合治疗B7-H3表达的复发或难治性肿瘤的安全性和有效性。总而言之,B7-H3是一种潜在的基于检查点的针对T细胞和NK细胞的免疫治疗的候选物。
自然杀伤细胞是一组独特的抗肿瘤效应细胞,具有不受MHC限制的细胞毒性、产生细胞因子和免疫记忆等功能,使其成为先天性和适应性免疫反应系统中的关键角色。一些癌症的发生与功能失调的NK细胞有关。因此,修复这种NK细胞可能是抗肿瘤免疫治疗的一个潜在选择。这种修复的一种方法是抑制免疫检查点,即癌细胞通过控制免疫细胞表面的抑制受体进行免疫逃逸。免疫检查点抑制在T细胞的情况下是成功的。NK细胞最近也被用于同样的目的。针对NK细胞表面的这些抑制受体的免疫检查点抑制剂,如monalizumab和lirilumab,已作为单药治疗进行,并显示出良好的安全性,但仅在延长无进展生存期方面取得了轻微成功。因此,免疫检查点抑制剂(如CTLA-4和PD-1抑制剂)的组合也可以在NK细胞的背景下尝试,因为抗PD-1和抗PD-L1抑制剂也被证明可以增强NK细胞介导的细胞毒性。同样,NKG2A增强肿瘤疫苗诱导的CD8 T细胞免疫也强调了联合治疗的潜力。因此,结合抗PD-1或抗PD-L1抑制剂和NK细胞特异性检查点抑制剂(如抗KIR或抗NKG2A抑制剂)可用于基于检查点抑制的联合免疫治疗。随着B7-H3、CD200R、CD47和Siglecs7/9等新的检查点的加入,将这些检查点结合起来进行协同抗肿瘤反应是未来充分发挥NK细胞杀伤肿瘤作用的方向。
参考文献:
1.NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol., 13 February 2020

版权声明/免责声明
本文为授权转载,版权归拥有者,仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系。衷心感谢!


推荐阅读
GLP-1的前世今生——口服GLP-1R激动剂与GLP-1赛道的展望(连载四) GLP-1的前世今生——GLP-1赛道的Best-in-class之争(连载三) GLP-1的前世今生——蜥蜴毒液带来GLP-1的开源治疗(连载二) GLP-1的前世今生—— 糖尿病与GLP-1的节流治疗(连载一) K药和O药双雄争霸、巅峰对决的故事 创新药时代,CMC先行!——新Logo,新海报!中国新药CMC高峰论坛全新驶来! 国家药监局药审中心副主任周思源:以临床价值为导向的药物研发与科学监管 国家药品监督管理局正式受理赛诺菲糖尿病创新药iGlarLixi上市申请 最强创投大会卫星会!双元盛锋Bio酒会连办六届,共议差异化创新赢得未来! HIV治疗正式进入基因编辑时代! 中国好BD | 阿诺医药 见证!“2020年度中国生物医药企业创新力百强”名单公布 光遗传学会不会获诺奖? 众望所归!2021拉斯克奖揭晓,两位mRNA先驱摘得桂冠,光遗传学也获奖了 灵魂拷问!如果mRNA技术今年能摘得诺奖,谁才是最大的贡献者? 国内药企走向合作“拐点”,恒瑞们离国际顶级药厂更近了? 讲座直播:2021年度诺贝尔生理学/医学奖花落谁家?| 药时代与返朴联合主办 创新药时代,CMC先行!——新Logo,新海报!中国新药CMC高峰论坛全新驶来! 腾盛博药——一个创新药领域的“四有青年”! 灵魂拷问!恒瑞应该对标哪一家外国药企?请投下您宝贵的一票! 报告!我要实名举报生物医药投资人中的两面派! 海和药物谷美替尼片获纳入突破性治疗药物品种用于治疗具有MET 14外显子跳变的局部晚期或转移性非小细胞肺癌 Moderna的成功难以复制?下一匹黑马会是谁? 热烈祝贺!恒瑞医药张连山、复宏汉霖张文杰、赛诺菲大中华区总裁贺恩霆等行业领袖荣获上海市“白玉兰纪念奖”表彰! 斗胆跨界说恒瑞 | 附:您认为恒瑞应该对标一家外国药企吗?请投票! 斗胆给恒瑞支支招! 辉瑞专栏 | 成功的CDMO合作如何为无菌注射产品的成功奠定基础 (第二部分,共计六部分) 深度雄文 | 重新定义恒瑞 中国罕见病定义研究报告发布:患病人数小于14万为罕见病 医药股的寒冬,离结束还有多远? 关注!莆田已现24例阳性,初判德尔塔!又一国产新冠“特效药”传来大消息…… 刚刚!2022科学突破奖公布,两位mRNA技术先驱与其他23名学者分享1575万美元奖金 中国仿制药辛酸往事:用量85%的仿制药,只花了12%的医药支出。人才怎样才能留在制造业? 3.8+万人观看!诺华人才空中宣讲精彩回顾 Promega开班在即!| 如此火热的PROTAC不可不知! 特别报道 | 明星公司今何在?十年过去了,这些生物技术公司还好吗?(下)







 点击这里,欣赏更多精彩内容!
点击这里,欣赏更多精彩内容!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权
 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 