

前期,我们推出了介绍正处于III期临床的ADC管线的系列文章。其中,在“中篇”中着重介绍的ARX-788利用合成氨基酸实现了抗体与Linker-payload的位点特异性偶联,提升了ADC产品的稳定性和临床治疗效果。由于同质性较高的ADC产品在药理学方面有着明显的优势,在通过选择性位点修饰制造高均一度的ADC产品方面已开发出多种策略。本期文章将跟随剑桥大学化学系发表的综述文章,深入ADC选择性位点修饰的进展情况。
ADC药物的偶联方式很大程度上受到抗体自身特性的影响。目前,已上市及在研的ADC产品都是基于免疫球蛋白(IgG)抗体的不同亚型为基础开发的。IgG共存在IgG1、IgG2、IgG3和IgG4等四个亚型,虽然他们具有约90%的序列同源性,但在血清稳定性、链间二硫键数量等方面存在差异。同时,由于其在通过抗体依赖性细胞毒性(ADCC)或补体途径激活免疫系统的能力方面存在差异,因此在ADC药物开发上有所侧重。
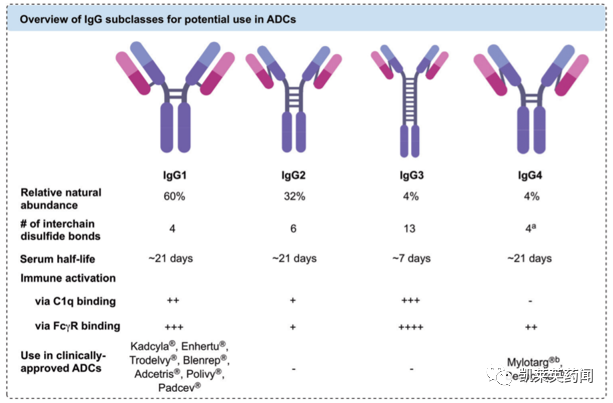
由于IgG1具有较长的血清半衰期和较强免疫激活能力,而在ADC药物的开发中被广泛使用。同时,在需要较少免疫激活的情况下,IgG4也被用于ADC药物的开发。IgG1和IgG4共含有16个二硫键,其中12个为链内键,4个为链间键。由于链间键高度暴露于溶剂中,其很容易通过化学方法减少或修饰。然而链内键埋在蛋白质的球状折叠中,因此对化学修饰不敏感。与IgG1相比,天然的IgG4分子可以进行Fab臂交换并导致其体内效力降低和无效的靶向效应。已上市产品Mylotarg和Besponsa均通过重链铰链区的S228P突变来预防上述情况的发生。
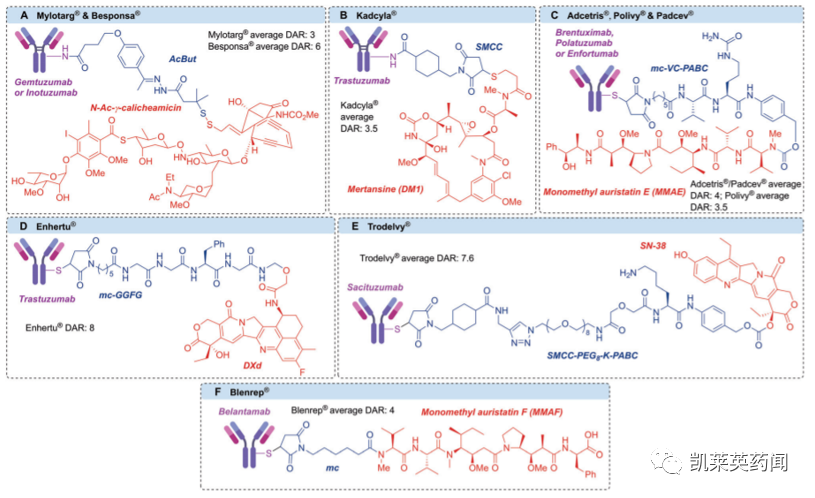
目前,针对Linker与抗体偶联的研究主要目标是提升抗肿瘤功效和增强药物安全性,主要在以下四个方面进行研发改进:
-
ADC偶联物在血液循环中保持高度稳定,避免有效载荷的过早释放;
-
-
-
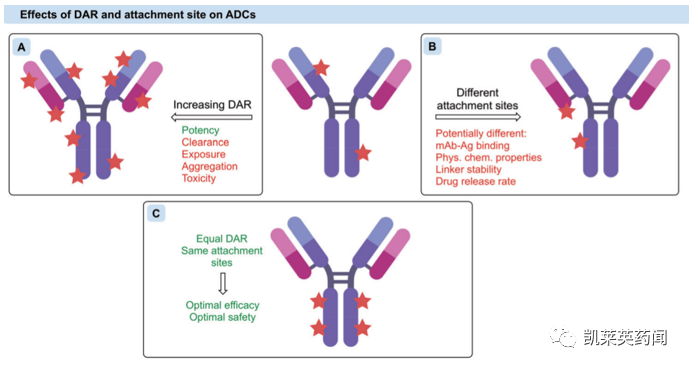
在载药量方面,虽然最大限度地增加DAR可以有效提高抗肿瘤效力,但由于常用细胞毒素往往是分子量较大的亲脂性物质,会导致蛋白质聚集和ADC清除率的增加,从而降低疗效和安全性。因此,平衡的DAR有助于在获得抗肿瘤效力的同时保持抗体的药代动力学特征。
除载药量外,偶联位点也是重要考虑因素。偶联部位应远离抗原结合区,使抗体结合和被细胞内化不受影响。此外,偶联部位还可以对偶联物的稳定性产生显著影响,后者直接决定了药物在体内循环和肿瘤部位中的释放速率。
综上,具有同质DAR和偶联位点的ADC可以产生更好的治疗效果。但鉴于IgG中存在大量反应性残基,在制剂研发过程中存在巨大的困难与挑战。近年来,位点选择性蛋白质修饰的进展使新一代ADC能够满足上述的同质性要求。
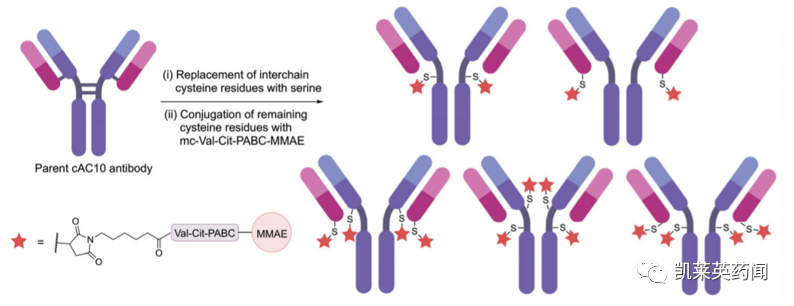
工程化半胱氨酸修饰(Engineered cysteines)
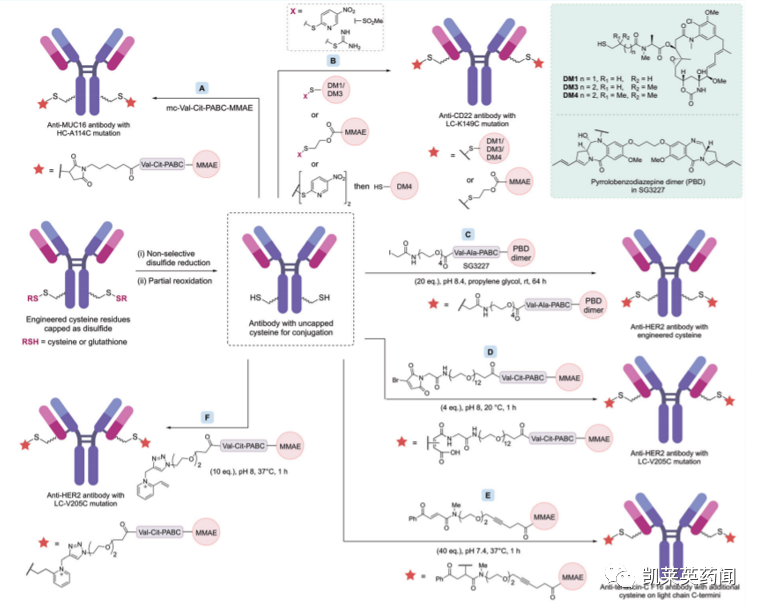
对抗体表面上可修饰的半胱氨酸残基进行工程化改造是实现位点选择性和均一性偶联的常用方法。最早实际应用方案是利用其他氨基酸对半胱氨酸残基进行替换,减少抗体链间的二硫化物的数量,从而保证可反应的半胱氨酸残基数量可控。如下图所示,通过用丝氨酸替换所选择的半胱氨酸,可以实现五种具有不同半胱氨酸位置的候选抗体,利用mc-Val-Cit-PABC-MMAE进行修饰,则可产生DAR为2或4的均质ADC
目前,除了上图所展示的通过马来酰胺(maleimides)进行抗体偶联外,基于工程化半胱氨酸修饰还发展出了基于直接二硫化物合成(下图B)、碘乙酰胺(iodoacetamides,下图C)等在内的多种均质ADC偶联技术。
二硫键重架桥(Disulfide rebridging)
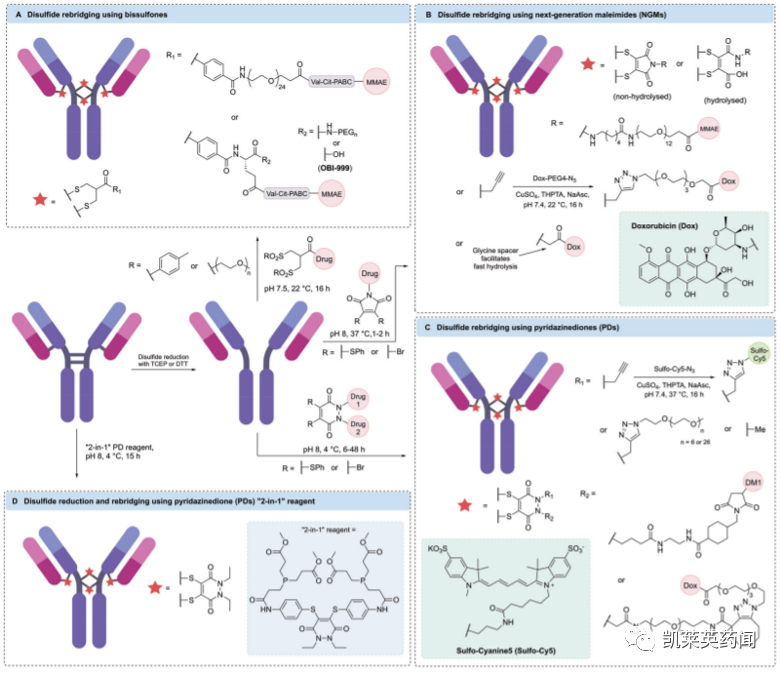
二硫键重架桥是利用半胱氨酸选择性交联试剂(如TECP或DTT),将IgG1抗体中4个链间二硫键还原,随后使用双反应试剂在多肽链重新连接的同时,完成小分子有效载荷的安装或抗体的进一步修饰。通过共价重新连接半胱氨酸残基,既维持了二硫键的稳定作用,同时实现每个二硫键链接一个有效载荷的受控偶联。根据链接子所携带的有效载荷数量,可获得DAR为4、8或16的ADC药物。
目前,根据所使用主流技术有双砜试剂(Bissulfones,下图A)、新一代马来酰亚胺(NGMs,下图B)、哒嗪二酮(PDs,下图C)、“二合一”PD试剂(下图D)等。
非天然氨基酸位点修饰(Non-canonical amino acids)
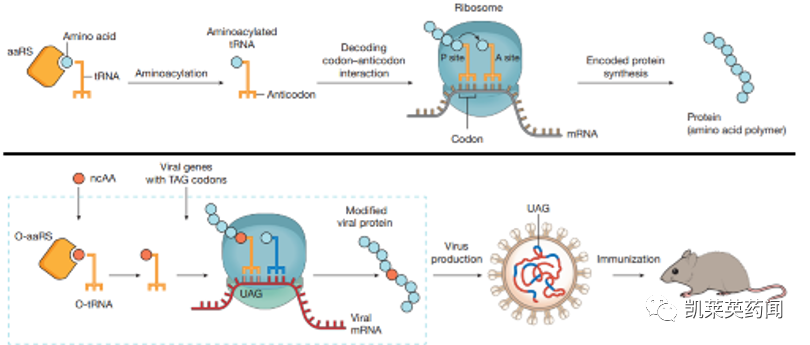
除了硒代半胱氨酸(Sec)等罕见氨基酸外,所有蛋白质都是由20种天然氨基酸合成的。近年来,随着遗传密码子扩展技术(GCE)的发展,可以有效地在蛋白质中引入非天然氨基酸(ncAA),从而实现位点特异性结合。在实际操作过程中,通过将可以识别并有效抑制终止密码子的tRNA和ncAA特异性氨酰tRNA合成酶(aaRS)导入到细胞内,从而制造在特定位点具有非天然氨基酸的抗体。
(上图为正常蛋白质合成,下图为在蛋白质中引入ncAA)
C-/N-端修饰(C-/N-terminal modifications)
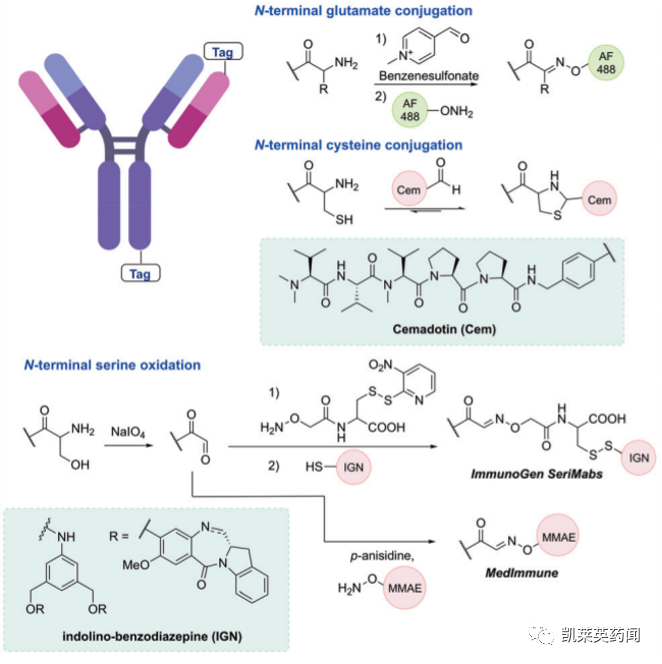
对抗体的N端或C端进行修饰已被证明是有效生产同质化ADC的可行策略,可以简单地在蛋白质链末端完成修饰。由于抗体C-端位置距离抗原结合区的距离较远,可以有效保护抗体结合的特异性和亲和力。与之相对,由于N-端距离抗原结合区距离较近,在进行N-末端修饰时需要注意其对抗体的抗原结合能力是否产生影响。
在实际应用中,N-末端转氨(N-Terminal transamination)是一种通过将N-端氨基转化为醛官能团,从而实现位点选择性合成ADC的策略。如下图所示,可以利用N-端的半胱氨酸、谷氨酸或丝氨酸等完成N-端修饰。
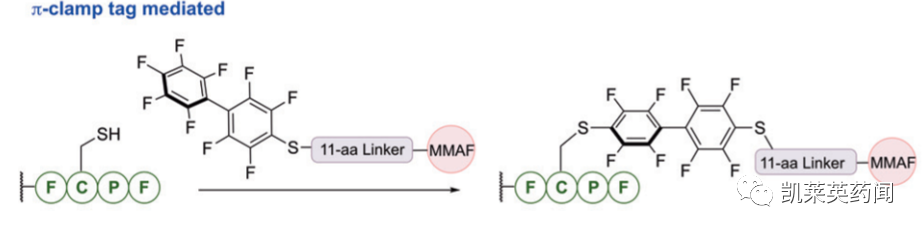
C-端修饰则可采用被称为π-clamp peptide sequence(FCPF)技术实现,可在存在竞争性半胱氨酸残基的情况下,用全氟芳烃试剂直接对半胱氨酸进行位点选择性修饰。
酶由于其高度的特异性和温和的反应条件,经常被用于实现位点选择性抗体修饰。酶可以直接将有效载荷连接到特定氨基酸序列,或者在抗体上引入反应性功能基团而后再实现有效载荷的连接。
谷氨酰胺转胺酶(Transglutaminase)
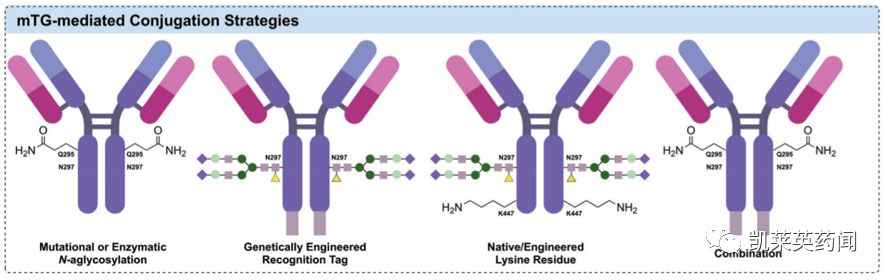
微生物谷氨酰胺转胺酶(microbial Transglutaminase,mTG)已被证明是可将有效载荷偶联到抗体上的酶促方法之一。mTG催化谷氨酰胺残基的γ-酰胺基和赖氨酸的6-氨基的酰基转移反应,形成分子内和分子间的肽交联。
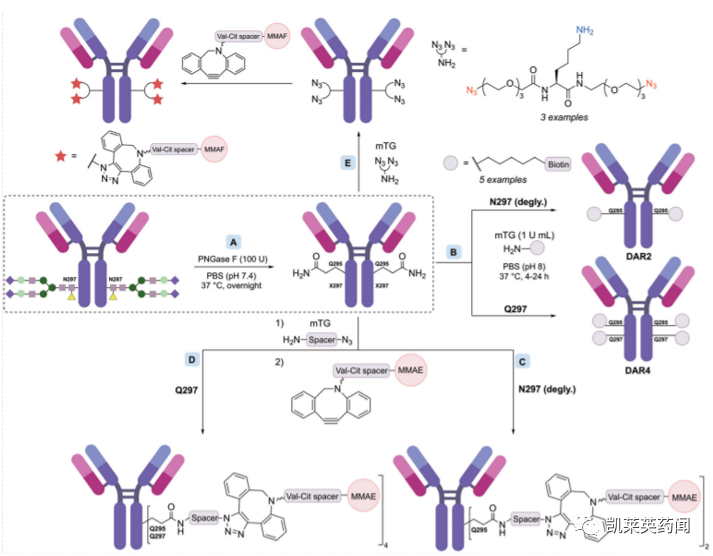
在实际操作中,为实现mTG介导的位点特异性修饰,可以通过酶促或突变等方式去除N-连接重链聚糖,从而暴露天然Q295位点的谷氨酰胺残基;或通过基因工程引入mTG特异性识别序列等方法。
其中,通过Q295位点可采取多种策略获得DAR为2或4的ADC产物。
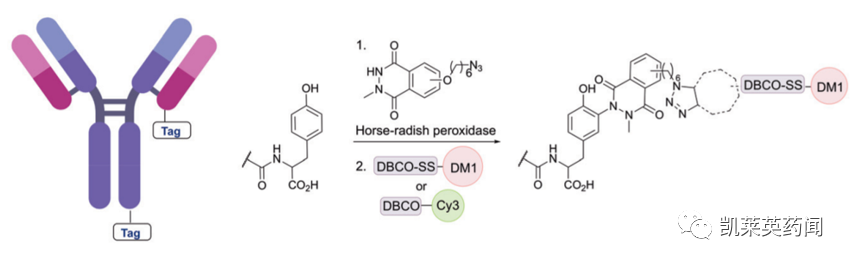
除mTG酶促修饰策略外,还可通过辣根过氧化物酶(HRP)催化酪氨酸酚(tyrosine phenol)的氧化策略,实现抗体C-/N-端的酶促修饰。
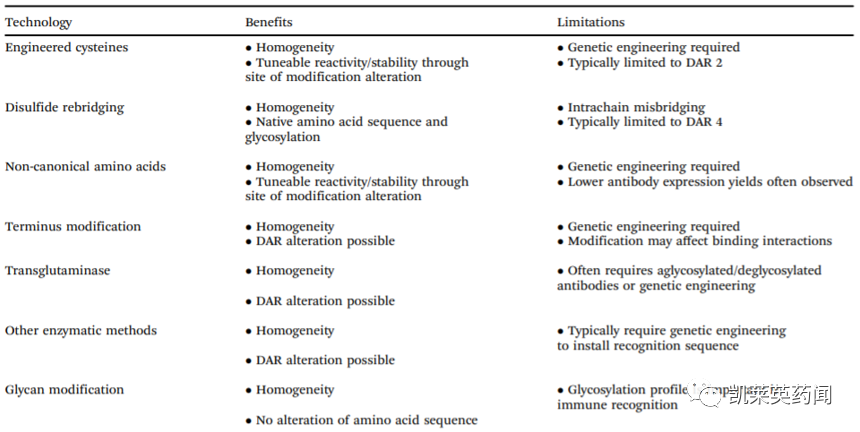
除上述化学修饰和酶促修饰外,还可通过聚糖修饰(Glycanmodification)等策略完成同质化ADC的生产。而无论何种策略,都具有其显著的优缺点(见下表):
目前,随着ADC药物在临床前及临床阶段的蓬勃发展,相信会有更多具有创新性的同质化ADC药物生产策略出现,为广大患者带来临床治疗效果更佳的药物选择。
1. Expanding and reprogramming the genetic code. Nature. 2017 Oct
2. Site-selective modification strategies in antibody-drug conjugates. Chem Soc Rev. 2021 Jan.
本文为授权转载文章,仅代表作者观点,版权归作者。
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!
 点击这里,与~20万同药们喜相逢!
点击这里,与~20万同药们喜相逢!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权



 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 