关注“药时代”微信公众号,获取更多精彩内容 ↓ ↓ ↓



晶研谈是由晶泰科技发起的药物固态研究交流专栏,旨在与学术专家及业界同行就药物固体形态研究中的问题与挑战进行深入交流,分享最新研究进展,讨论前沿创新技术,探索有效解决方案,共同促进国内药物固体形态研究的发展及繁荣。
相同的药物分子因其晶型不同而具有不同的理化性质,而理化性质的不同决定了药物在人体内的生物利用度,生物利用度又最终影响药效、给药形式以及计量等问题。
因此在现代药物研发的临床期研究中,进行完备的晶型筛选实验是非常重要的一个环节。但单纯地通过实验进行晶型筛选是投入时间长、人力成本高,且没有明确终点的事情。换句话说就是——即使已经通过实验发现了多种晶型,依旧无法判断是否还存在其他稳定晶型!
以美国雅培在 1996 年上市的抗艾滋病特效药利托那韦为例,它上市时是以晶型I存在的;但两年后即 1998 年市面上在售的利托那韦被检测出存在晶型II,因此雅培又重新进行了晶型研究;而仅仅四年后(2002年) TransForm 公司却又发现了它的第三种晶型!
通过计算机模拟来进行或辅助药物晶型筛选,一方面可以缩短实验周期;另一方面可以给出晶型研究是否完备的理论预估。随着计算机算法和算力的快速发展,自 2016 年以来,晶型预测技术就被逐步深入应用到药物研发流程的各个环节中。
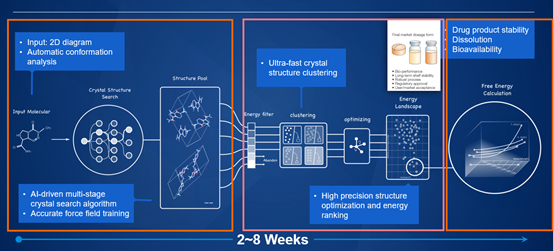
晶型预测(Crystal Structure Prediction,简称CSP)是指给定分子的二维结构式通过计算模拟获得它的所有可能的稳定晶型。CSP 流程共包含三个主要阶段,晶体搜索、能量排位和室温稳定性计算。其中每个阶段都包含一些关键难点,接下来会详细介绍。
晶体搜索在数学上表达为一个高维空间的全局优化问题。即构造一个关于晶体所有自由度的能量函数,求解这个能量函数的全局最小点和一部分局域极小点的问题。晶体自由度包含分子的内禀自由度、晶胞参数、分子在晶胞中的位置,以及晶体的对称性即空间群类型。其中分子内禀自由度包括柔性二面角的个数和环异构数目以及可能的手性;晶胞参数包含三个晶格常数和三个晶轴夹角;分子在晶胞中的位置包含分子在晶体坐标系中的质心坐标和分子相对于坐标系的方向;而晶体对称性可以是自然界中 230 个空间群中的任意一种。而搜索问题的计算复杂度还随着晶体自由度的增加呈指数级增长。
针对这样一个困难的高维优化问题,我们采用了三种主要的解决思路:首先是随机算法和启发式算法结合的全局优化算法,随机算法采用并行蒙特卡洛,这个算法在数学上是可以严格证明当采样充分时总能到达全局最小点的,但它的问题在于采样效率低,因此这里还结合了粒子群算法这类启发式算法提高搜索速度和效率;其次是使用随机和局域优化结合的结构生成算法,这是因为复杂体系随机生成结构生成效率低,因此采用随机生成加局域优化来提高结构生成效率,能够将预先知道的化学信息和规则加入局域优化器,使得生成化学合理的虚拟结构比例大幅度提高;第三是引入机器学习算法主动识别低能区并增强采样,进一步提高搜索效率。通常在晶体搜索阶段能得到数百万甚至千万量级的具有化学合理性的虚拟晶体结构。
通常来说,量子化学精度的计算可以得到较为可靠的相对稳定性和晶体结构,但搜索阶段产生的虚拟结构数量级过大,如果全部使用量子化学精度的计算则非常昂贵且耗时,且常规精度的量化计算的时间复杂度随体系复杂度呈 O(N3)~O(N4) 增长,当体系过于复杂之后,这就成了不可解决的问题,更不用说高精度的量子化学计算了。而另一方面,基于经典力学的力场方法速度快,但它与量化能量相关性不高。所以在实际计算中,我们采用多轮精度不同的排位策略逐步筛选的方法,使得 CSP 在药物工业中变得真正实用。多轮精度不同的排位策略可以逐步筛除掉部分高能结构,从力场到半经验方法可以将结构数从百万减少到万或者十万量级,从半经验方法到量子化学计算,可以进一步将结构数减少到千量级。最终阶段只对极少数结构进行高精度量子化学计算,将相对能量误差控制在 1.5kJ/mol 以内。
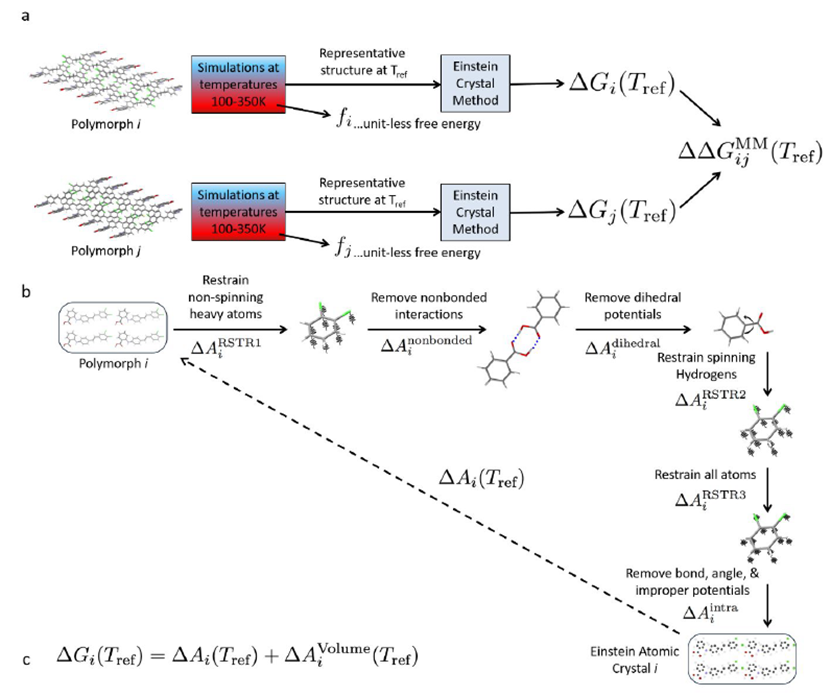
量子化学计算的是晶体在绝对零度下的能量,但实际上药物研发中所需要考虑的晶体稳定性通常是室温稳定性,这就需要进行有限温度的自由能计算。这里我们采用两种不同框架的计算在低温段互相验证,确定最终的室温稳定性。基于简谐近似或准简谐近似的晶格动力学理论,可以通过计算声子态密度的积分来获得绝对零度附近的任意低温区的赫姆霍兹自由能,从赫姆霍兹自由能可以判断有限温度的稳定性,这种方法是基于量子的,通常认为精度较高,但由于它的理论基础是准简谐近似,所以计算结果只在低于 150K 的低温下可靠,温度稍高一点结果就不再可信了。所以实际计算室温 300K 的稳定性就需要用到基于严格热力学循环和构型采样的准超临界路径方法 PSCP(pseudo super-critical path)【1】。这个方法需要用到假想的爱因斯坦晶体,某一个晶体结构它对应的爱因斯坦晶体是指跟它的原子的空间三维坐标一致,但去掉全部相互作用的虚拟体系。当比对两个化学成分相同但构型不同的晶体结构时,只需要对这两个结构分别构建热力学过程从有相互作用的状态逐渐去相互作用变到他们各自的爱因斯坦晶体态,那么这两个结构的自由能差等同于他们分别变化到各自的爱因斯坦态的自由能变化的差。这样就避免了采用分子动力学模拟计算一个晶体升华过程绝对自由能由于构型变化导致的采样不充分问题,可以用较少的模拟时长就能得到两个晶体之间较精确的自由能差。但 PSCP 方法的结果可靠性非常依赖力场参数对动力学过程中的结构描述的准确性,所以在实际应用中我们通常会拟合多版专有力场参数,并将它们的低温段(<150K)的分子动力学模拟自由能趋势与前面介绍的基于谐振近似的晶格动力学自由能趋势进行对比,选择二者符合较好的一版力场参数,使用 PSCP 方法计算室温下的自由能。
接下来从具体例子介绍晶型预测(CSP)在药物研发中的 3 个主要应用场景:
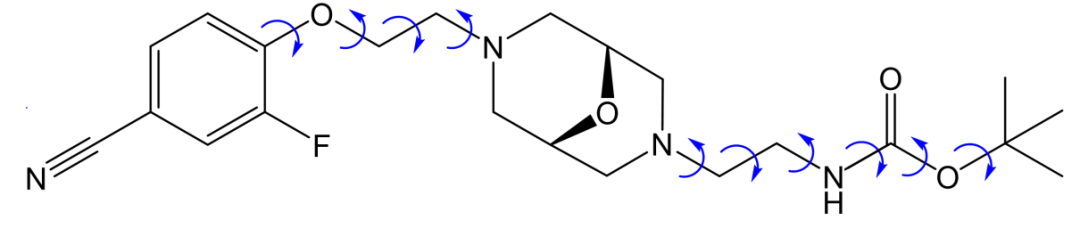
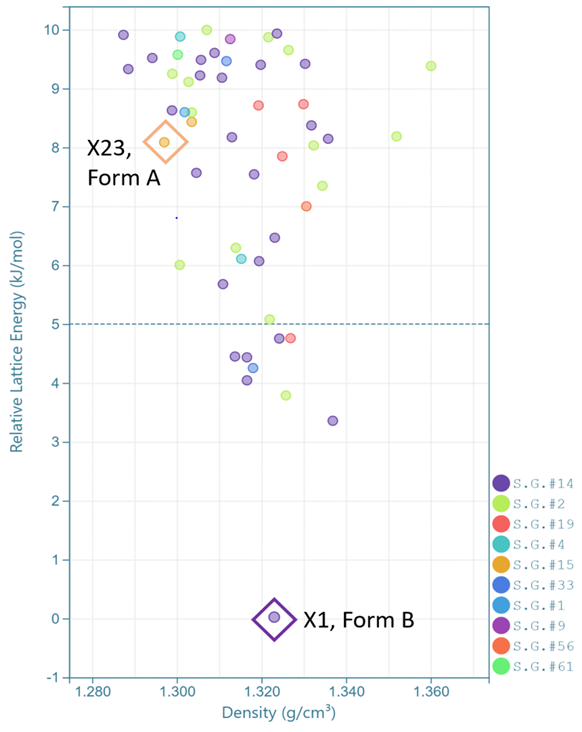
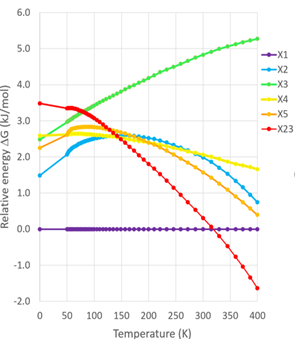
这是晶泰科技和美国阿斯利康合作研究的一个工作【2】。AZD1305(图4)是一种治疗心血管疾病的分子,通常这样柔性自由度高、分子复杂的体系,它的多晶型现象会很复杂,晶型之间的相对稳定性情况也会比较复杂,阿斯利康通过实验筛选到了两种晶型,但是不确定筛选是否完备,即是否还有可能存在其他更稳定的晶型。于是晶泰科技药物固态研发团队做了 CSP 研究,并成功地找到了两种实验晶型 Form A 和 Form B,在 landscape 中对应的虚拟晶型分别为 X1 和 X23 。此外,自由能计算证明室温下(300 K)Form A 和Form B是两种最稳定晶型,其他虚拟晶型的室温稳定度均不如这两个,理论计算(图5,6)基本上排除了室温下存在其他更稳定晶型的风险。
2. CSP 第二个应用场景是指导实验制备新晶型,比如推荐结晶条件。
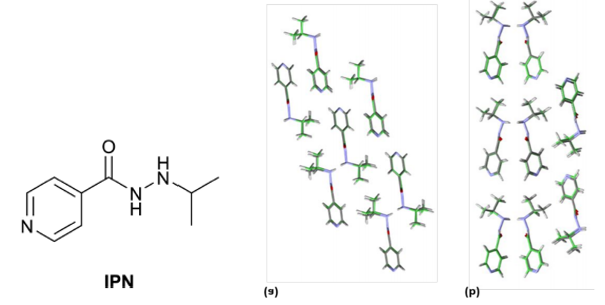
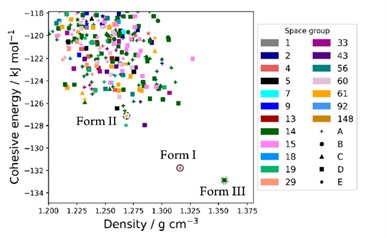
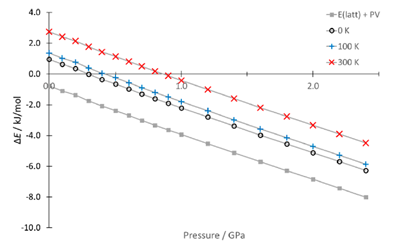
这是一个通过理论计算的信息帮助实验选择非常规条件,获得异丙烟肼最稳定晶型的例子【3】。大量常规实验结晶包括溶液相结晶、凝胶相结晶、升华结晶等方法都只能得到 Form I 和 Form II。但 CSP 发现在晶格能低于 Form I 的区域还存在一个虚拟晶型,那虚拟晶型有一定可能性是能真实存在的。从能量-密度图(图8)中观察到这个结构的密度大于所有其他结构。进一步进行加压晶格能计算发现这个结构随着压力增加晶格能持续下降(图9),且始终低于 From I。综合密度大和加压稳定性增加两方面的信息,猜测高压可能可以稳定这个虚拟晶型。随后的高压结晶实验确实证实了理论预测的结果,在 Merrill−Bassett diamond anvil cells DACs(diamond anvil cells)中压缩常压下长出来的晶体,当压力增加到 2.1 GPa 时,实验观察到 Form I 发生了转晶,通过单晶X射线衍射证明新发现的晶型确实为 Form III。(本案例来自文献报道,晶泰科技与药企合作加压结晶预测的案例由于保密性要求没有在这里展示)
图7 异丙烟肼(左), Form I (中), Form II(右)
3. CSP 的第三个用途是药物发现早期的溶解度预测
由于溶解度直接影响药物的生物利用度并最终影响药效,因此在药物发现早期,获得溶解度信息对于先导化合物的选择和优化都有重要意义。但由于处在药物发现早期,先导化合物数量大,如果全部使用实验方法精确测定溶解度,则面临合成任务重,合成困难以及周期长的问题。因此如果通过计算在不需要合成的时候能够又快又准地得到溶解度则能直接加速药物研发过程。但它的一个较大的困难是先导化合物分子之间较相似,在没有晶体信息的情况下通过理论预测这些相似分子的溶解度区分度不高。
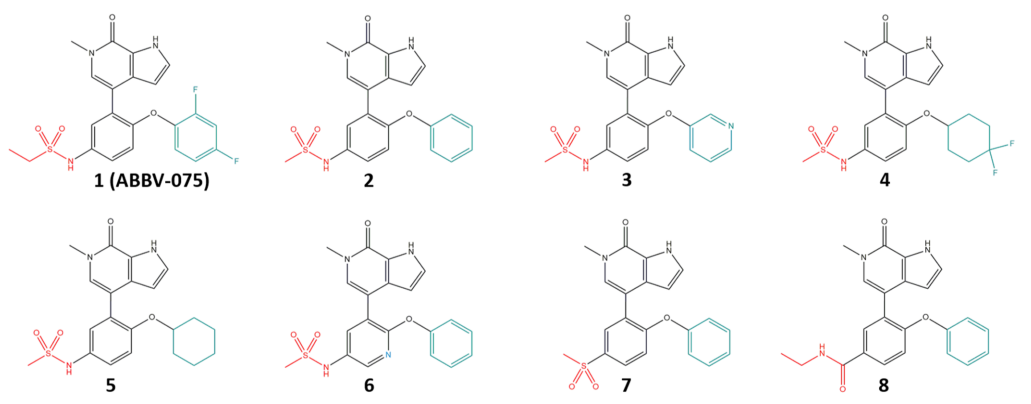
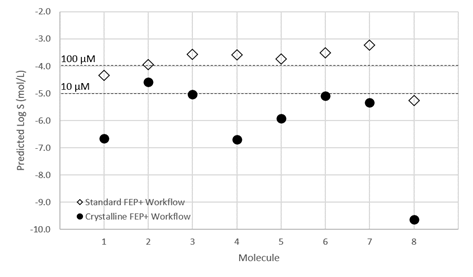
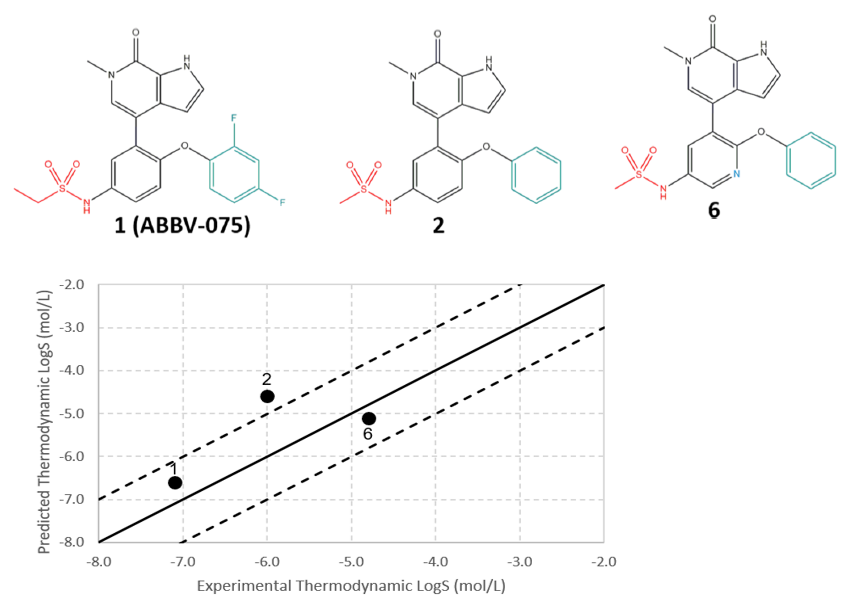
这里展示的例子是晶泰科技和美国 AbbVie 公司合作研究的一个早期溶解度预测的工作【4】,文章中针对 AbbVie 早期设计出的 8 个相似分子(图10)进行了溶解度预测。传统的溶解度预测,无论是 QSAR 方法,还是基于无定型态构建热力学循环计算溶解自由能的方法,由于都无法将晶体堆积作用对溶解度的影响考虑到模型中,因此预测出来的溶解度数据很难区分开具有相似骨架的同系列分子,如图 11 中的白色空心方点所示;但基于晶体结构信息的溶解度预测数据在相似分子间更具有区分度,如图 11 中黑色圆点所示。且和已有的热力学溶解度数据对比,预测溶解度和实验溶解度的误差更小(图12),因此可以用作先导化合物筛选的信息。最终, Abbvie 根据我们的溶解度预测值和其他 ADMET 值一起进行综合考虑,选择了 8 个分子中的一个进行后续开发。
图11 基于无定型态(空心方点)和基于晶体(黑圆点)的溶解度预测
综上所述,晶型预测(CSP)在技术层面可有效评估现有晶型间相对稳定性,及当前晶型研究的合理性,从而更为精准地评估药物后期的转晶风险并加速决策;在战略层面可评估现有晶型研究是否完备,从而对于药企知识产权布局及药品全生命周期管理提供可靠的依据。
CSP 技术无需实验数据辅助,因此可根据各类型创新药项目的推进计划灵活选择研究时间。同时也因其高效、准确的服务质量,众多国内外顶级药企已将 CSP 纳入小分子药物研究的标准流程。在过去几年里,晶泰科技已通过 CSP 技术加速了客户 100 多款创新药的开发,经过5年的经验积累和精益求精的技术迭代,近 3 年 CSP 预测的准确率始终保持在 100%。
未来我们会进一步优化 CSP 的算法,缩短周期并降低成本;同时结合 AI 算法,进一步精准推荐结晶条件,致力于为国内外客户提供更为优质的固研服务。
如果您有相关问题,欢迎给本篇文章「留言」或点击文末「阅读原文」按钮联系我们,期待与您进一步交流与探讨。业务咨询请联系:bd@xtalpi.com
[1] Cryst. Growth Des. 2020, 20, 8, 5211–5224
[2] Cryst. Growth Des. 2021, 21, 4, 1972–1983
[3] Taylor, C.R., Mulvee, M. T. , Perenyi, D. S. , Probert, M. R. , & Steed, J. W. . J. Am. Chem. Soc., 2020
[4] J. Chem. Inf.Model. 2021,61, 3, 1412–1426
晶泰科技是一家量子物理与人工智能赋能的药物研发公司,通过提高药物研发的速度、规模、创新性和成功率,致力于实现药物研发的行业革新。作为一家立足中美、服务全球的企业,晶泰科技始终坚持探索最优解决方案,以充分利用前沿的研发与计算资源,最大化满足客户与合作方的需求。
晶泰科技的智能药物研发平台将基于云端超算数字化研发工具与先进的实验能力进行整合,形成高精度预测与针对性实验相互印证、相互指导的研发系统。作为全球先锋人工智能药物研发公司之一,晶泰科技已建立起一整套量子物理干实验室与先进湿实验室紧密结合的研发迭代流程,挑战传统研发的效率瓶颈,赋能新药研发实现创新速度与规模的突破。
本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权




 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 