今天下午3点,杜博士解读《创新时代大潮来临,中国迎来新药收获期》


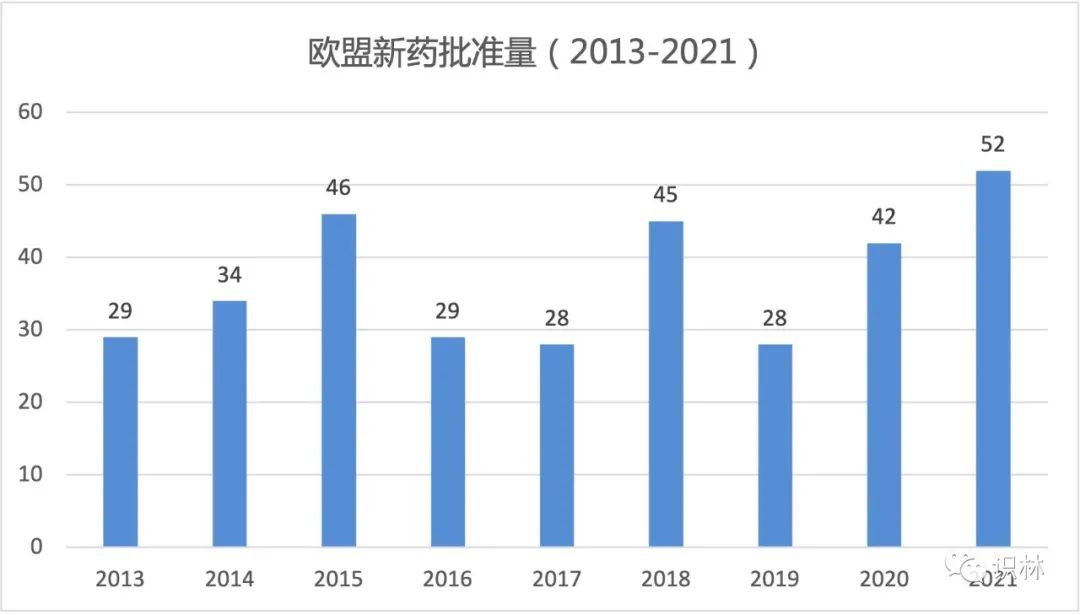
2021 年欧盟批准了创纪录的 52 个含有新活性成分(NAS)的药物,其中包括批准了用于新冠疾病的 4 个新疫苗和 3 个新药。
与 2020 年(42 个含 NAS 的产品)相比,该数字增加了近四分之一,比 2019 年(28 个)增加了近一半。过去十年中,创下批准最多记录的一年是 2015 年,有 46 个含 NAS 新药获得上市许可(MA)。
虽然新冠疫情加剧了欧洲药品管理局(EMA)已经迅速增加的工作量和资源问题,但 EMA 受益于引入的一些监管灵活性,以允许更快速地开发、评价和批准新冠疫苗和药物,其中包括滚动审评、更快批准流程以及有条件上市许可的使用。
与 2020 年一样,2021 年批准了 17 个罕见病药物。其中包括 Deciphera Pharmaceuticals 公司用于治疗晚期胃肠道间质瘤的 Qinlock (ripretinib),Incyte Corporation/MorphoSys AG 公司用于治疗弥漫性大 B 细胞淋巴瘤的 Minjuvi (tafasitamab),以及罗氏用于 5q 脊髓性肌萎缩症儿科患者的 Evrysdi (risdiplam)。
另外批准了 17 个用于肿瘤适应症的新药,其中一个是新基/百时美施贵宝公司用于成人复发和难治性多发性骨髓瘤患者的基因疗法 Abecma (idecabtagene vicleucel)。
另一获批上市的基因疗法是 bluebird bio 公司用于早期脑型 X 连锁肾上腺脑白质营养不良症的 Skysona (elivaldogene autotemcel)。
值得注意的是,去年批准的产品之一包含两种新活性成分:罗氏/再生元的新冠治疗药 Ronapreve (casirivimab + imdevimab) 。而另外两个新药,口服避孕药 Gedeon Richter 公司的 Drovelis 和 Estetra 公司的 Lydisilka 含有相同的 NAS — 雌四醇(与屈螺酮组合)。
17 个肿瘤药批准之一是阿斯利康公司用于成人复发或难治性毛细胞白血病患者的 Lumoxiti (moxetumomab pasudotox),该产品是在特殊条件下获得批准的孤儿药。上市许可在获批 5 个月后因商业原因应申报人要求撤回。
2021 年,14 个新产品在欧盟获得有条件上市许可(CMA)。在 CMA 下获得许可的申办人同意提供更多产品数据,以期最终转为完整的上市许可。
去年的 CMA 包括四个新冠疫苗:Moderna 公司的 Spikevax,AstraZeneca/牛津大学的 Vaxzevria,强生新冠疫苗,以及 Novavax 的 Nuvaxovid。欧盟首个新冠疫苗辉瑞/BioNTech 的 Comirnaty 已于 2020 年 12 月获得批准。
2021 年,欧盟还根据标准上市许可程序批准了 3 个含有 NAS 的新冠治疗药:罗氏/再生元的 Ronapreve,Celltrion 的 Regkirona (regdanvimab)以及葛兰素史克/Vir 公司的 Xevudy (sotrovimab)。
另外,还有两个现有用于炎症性疾病的药物 — 瑞典 Orphan Biovitrum AB 公司的 Kineret (anakinra)和罗氏/Chugai 公司的 Actemra/RoActemra (tociluzumab),将欧盟适应症扩展到包括治疗严重新冠疾病患者。
4 个新药受益于另一欧盟监管机制:特例批准 , 这 4 个药分别是:Stemline Therapeutics 公司用于母细胞性浆细胞样树突状细胞肿瘤的 Elzonris (tagraxofusp),再生元用于纯合子家族性高胆固醇血症的 Evkeeza (evinacumab),Albireo Pharma 公司用于治疗进行性家族性肝内胆汁淤积症的 Bylvay (odevixibat) , 以及批准后撤回的 Lumoxiti。
特例批准用于因为所讨论的疾病很少遇到或者收集信息是不道德的,从而在正常使用条件下不能期望申办人提供关于其产品的全面安全性和有效性数据的情况。
两个孤儿药 Bylvay 和 Evrysdi 于 2021 年在加速程序下获得批准。欧盟加速审批用于对公共卫生具有重大意义的药物 , 审评时间为 150 天,而不是标准的 210 天。
欧盟新临床试验系统将于月底上线,申办人应注意哪些问题?
欧盟新临床试验信息系统(CTIS)将于 2022 年 1 月 31 日上线,届时欧盟临床试验的实施和监督将发生根本性转变,为临床试验法规(第 536/2014 号)的生效铺平道路。
CTIS 由欧洲药品管理局(EMA)开发,旨在帮助实现 2014 年 4 月临床试验法规通过时所承诺的诸多好处,例如,协调临床试验申请的提交和评估,提交试验透明度。对于制药企业来说,CTIS 的推出会带来很多新的实践和运营考虑。
首先,企业必须清楚地了解如何在商业利益与新的临床试验透明度要求之间取得平衡。这是因为除非同意延期,CTIS 的默认设置是在第一时间公开几乎所有作为临床试验申请的一部分提交的数据和文件(商业机密和个人信息除外)。
根据 EMA 的 CTIS 专家 Laura Pioppo 的说法,这意味着当申办人在 CTIS 中创建新的临床试验申请时,将必须提交大多数文件的两个版本,例如,试验方案、研究人员手册以及研究用药品卷宗的安全性和有效性部分。一个版本将向“公众”发布,这个版本不应包含任何个人数据。另一个“非公开”版本将仅对监管机构可见,并且可以包括姓名和签名或其他个人信息,以支持试验的评估。
她补充表示,唯一永远不会公开的文件是那些与质量(及质量评价)以及研究人员和试验场地之间的财务安排有关的文件。
计划制定战略以平衡透明度要求与商业利益的公司需要尽早开始准备,因为在提交初始申请时必须做出许多关键决策,例如:
-
将“公开发布”和“非公开发布”版本的临床试验文件同时上传。
-
行使选择权,延迟发布用于“公开发布”的文件以及基于正当理由提交给 CTIS 的某些结构化数据(例如,试验标题、试验设计、治疗意图等)。所有此类延期请求都将由相关成员国审查,如果其不同意申办人的提议,可能会要求提供更多信息。延期时间表将取决于试验类别,欧盟/欧洲经济区允许进行 1 类试验(首次人体试验,PK/PD、BE/BA、生物类似性研究)的最长期限为试验结束后七年。
Pioppo 解释指出,申办人以后不能再对这些关键决定进行更改。例如,如果在提交初始申请时没有提出延期请求,那么所有文件/数据将在对试验申请作出决定后立即公开。她澄清指出,在多国试验的情况下,文件/数据将“在第一个成员国做出发布决定后”公开,而无论报告成员国是否仍在审查数据。
Pioppo 表示,在检测环境中使用 CTIS 的虚拟版本时,试验许可后到允许公众访问研究文件/数据之间的时间间隔为 15 分钟。CTIS 于 1 月 31 日上线后,Pioppo 表示,“我无法确定是 15 分钟还是 30 分钟,但预计不会需要几天时间。”
Pioppo 表示,延期设置必须在一开始就达成一致。AbbVie 的 Scott Feiner 表示,当申办人要求延迟发布文件/数据时,不必证明延迟本身是合理的,而是必须解释他们的试验在临床试验法规下是如何分类的,从而使他们有权要求延期。例如,在 1 类试验的情况下,如果申办人要求推迟公布试验的主要特征,CTIS 中有个额外的方框来证明推迟的合理性。这一合理性说明将与研究人员手册一起发布。
在透明度和商业利益上取得平衡只是公司在准备临床试验法规合规时必须处理的一个方面。他们还需要了解 CTIS 的工作原理,CTIS 是一个由提交门户和数据库组成的高度复杂的 IT 系统。
在 CTIS 中,用户可以被赋予与特定项目或组织账户相关的不同角色和职责。为有效地使用 CTIS,公司被告知要找到在规定期限内执行各种与试验相关的任务所需的用户数量的适当平衡。EMA 提醒企业多个用户会使系统负担过重,并且创建多个备份用户会危及用户管理。另外,CTIS 还链接到几个欧盟“主”数据库来提取信息,例如 EMA 现有的组织管理系统数据库,以及 EudraVigilance 医药产品词典(xEVMPD)。
为帮助试验申办人和成员国熟悉该系统,去年 10 月份,EMA 提供了 CTIS 培训环境。目标是,“帮助未来的用户及其组织探索用户配置以及提交和授权临床试验申请所需的步骤。”EMA 补充表示,“测试环境仍然可以使用,并且在 CTIS 上线后仍可使用。其他申办人可能会在适当的时候获准进入培训环境。”EMA 还开发广泛的在线模块化培训计划和申办人手册,以帮助申办人做好准备。
为帮助企业适应新的工作方式,临床试验法规有个三年的过渡期 , 不过还是鼓励申办人尽早采纳 CTIS 系统。
-
在第一年(直至 2023 年 1 月 31 日),申办人可以选择根据现有的欧盟临床试验指令(通过 EudraCT)或根据临床试验法规(通过 CTIS)申请新的试验许可。但所有成员国必须从第一天开始就准备好使用 CTIS,并仍运行旧流程。
-
在第二年(从 2023 年 1 月 31 日起) , 根据临床试验法规提交成为强制性要求,申办人必须通过 CTIS 提交所有新申请。
-
到第三年年底(2025 年 1 月 31 日) , 根据临床试验指令批准的所有正在进行的试验都需要过渡到临床试验法规,并通过 CTIS 进行管理。

本文为授权转载文章,仅代表作者观点,版权归作者。
仅供感兴趣的个人谨慎参考,非商用,非医用、非投资用。
欢迎朋友们批评指正!衷心感谢!
文中图片、视频为授权正版作品,或来自微信公共图片库,或取自网络
根据CC0协议使用,版权归拥有者。
任何问题,请与我们联系(电话:13651980212。微信:27674131。邮箱:contact@drugtimes.cn)。衷心感谢!
 点击这里,与~20万同药们喜相逢!
点击这里,与~20万同药们喜相逢!本篇文章来源于微信公众号:药时代
发布者:药时代,转载请首先联系contact@drugtimes.cn获得授权


 为好文打赏 支持药时代 共创新未来!
为好文打赏 支持药时代 共创新未来! 